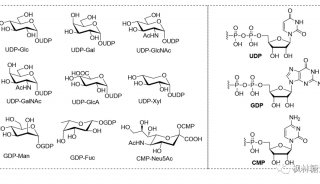
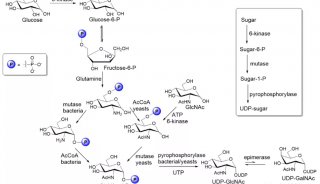
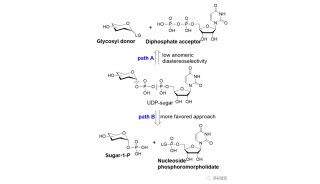
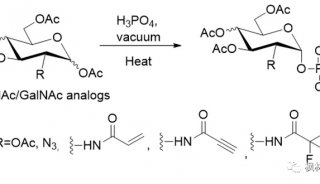
发文至

 分享至
分享至



分享至
广州生物院实现天然产物(-)-Terpestacin的不对称全合成
萜类天然产物(−)-Terpestacin可以抑制导致HIV病灶的细胞合胞体(syncytia)的形成(ID50 0.46 µg/mL),同时也能抑制血管增生作用,而且其生物活性具有较好的选择性。该化合物被认为是一种非常有前景的抗癌和抗HIV的先导化合物。
(−)-Terpestacin的主体结构是五员环和十五员大环反式稠合(即[3,0,13]双环骨架体系)而成的萜类化合物,大环上有三个反式三取代双键。共有四个手性中心(即C1, C11, C15, C23),其中C1位是季碳手性中心。五员环为官能团密集的1,2-双酮结构,其中一个羰基呈现烯醇式结构。以上结构特征使得该分子进行全合成具有较大的挑战性。鉴于上述生物活性和结构特点,该分子已成为许多世界著名大学(包括哈佛大学、斯坦福大学、麻省理工学院等)的著名学者的研究对象。目前文献中已有六个研究组完成了该分子的合成(含四条不对称合成路线),但合成步骤较长。
中科院广州生物医药与健康研究院邱发洋实验组以廉价的商业原料(R)-Carvone和(E,E)-Farnesol为起始原料,实现了 (−)-Terpestacin的全合成。在构建该分子的关键位点如1位的季碳手性中心、11位手性羟基和23位的手性甲基时,所用方法巧妙简洁,使全合成效率大大提高。
该合成路线仅涉及简单试剂和常规反应,便于实验室较大规模合成,为针对该化合物的进一步结构改造奠定基础。
相关论文已经发表在Organic and Biomolecular Chemistry杂志上。
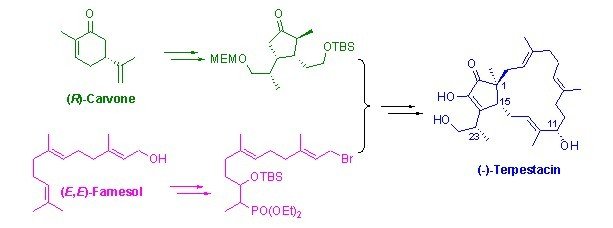
反应过程图
推荐
-
科技前沿
-
焦点事件
-
科技前沿
-
焦点事件

-
项目成果
-
焦点事件