
查看完整版本请点击这里:
【求助】Ni柱纯化蛋白如何提高结合率?含图
【求助】Ni柱纯化蛋白如何提高结合率?含图
如下图,我用的是novagen的His bind resin,没有过柱,按照操作手册上的方法在EP管里进行小批量的纯化,在4度翻转结合了1h,这里的上样量大约相当于500ul的菌液。从这个图的结果来看,似乎大部分蛋白都没有结合上ni柱,请问要如何解决这个问题??我的结合、漂洗、洗脱溶液的咪唑浓度分别是10mM、60mM、1M,好像很多蛋白在漂洗阶段就被洗下来了,要怎么办呢?看到有帖子推荐用不同的咪唑浓度进行梯度洗脱,这样有什么好处?我直接用1M的咪唑浓度理论上应该是都洗脱下来了啊,有必要做梯度吗?请多多指教!!
查看完整版本请点击这里:
【求助】Ni柱纯化蛋白如何提高结合率?含图
【求助】Ni柱纯化蛋白如何提高结合率?含图
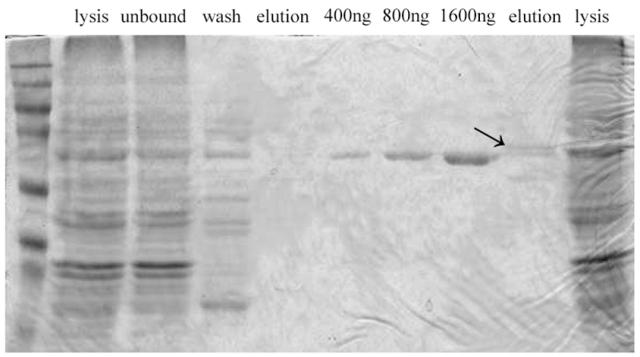
20121191.snap.jpg

最新回复
chengjie79 (2014-1-18 15:58:44)
还有一个问题,我的目的蛋白底下还有杂带,怎么除去这些杂带呢?因为我的蛋白为了助溶在N端还接了GSTtag,整个蛋白70多KDa,是不是发生了断裂啊?
yjf1026 (2014-1-18 15:59:06)
看上去那个 1600 ng 的洗脱条件不错啊。
chengjie79 (2014-1-18 15:59:39)
glass (2014-1-18 15:59:57)
感觉表达不是很好嘛。。你那目的条带在啥位置哇?应该在MARKER的哪个位置啊?
remenb (2014-1-18 16:00:19)
1 结合的时候加大样品与resin的比例(使用10ml菌液)。
2 漂洗咪唑浓度太高了,建议从20mM开始洗涤。100、200、 500mM进行洗脱,同时检测你的目的蛋白。这样同时可去除不必要的杂代。
bamboo16 (2014-1-18 16:00:37)
ukonptp (2014-1-18 16:00:56)
先确定有效表达,其次再来做纯化
langlang (2014-1-18 16:01:16)
500ul菌液太少了!建议1mL 菌液,收菌后溶到100uL lysis buffer中,与20uL填料结合,结合时间一般10min足够。lysis buffer 不要含imidazole,然后按50 100 200 300 400 500mM imidazole的梯度洗。提高pH和上样量有助于提高结合能力。有杂带的话考虑下一步纯化吧。
chengjie79 (2014-1-18 16:01:36)
目的条带就是箭头指的那条!
我实际操作中是将35ml的菌液用bind buffer 重悬,加入溶菌酶、超声,然后和70ul的resin 结合1h的。所以大概是 1ml 菌液:2ul resin的比例,加大比例的话是指多用一些菌液吗?
我的蛋白是有表达的,用wb 也已经检测到了。可能可溶蛋白产量确实不高,不知道大家有什么高招提高表达量呢?
liuaishan 说“lysis buffer 不要含imidazole”,为什么?如果我不用bind buffer来重悬的话,裂解液在纯化时要怎么换成bind buffer呢?还是说不用换?
感谢诸位的指点!!
sunnyB (2014-1-18 16:01:59)
首先做WB定性鉴定,做ELISA测表达量做初步判断,确定的确有表达而电泳的加样量可以看到条带。如果确定纯化时没有结合再优化纯化条件。
你目前的binding buffer的组分是怎样的?pH是多少?你把纯化条件传上来我再帮你调整。
chengjie79 (2014-1-18 16:02:28)
我又做了一次纯化,按照大家说的做了一下咪唑浓度梯度,我用得是novagen的His•Bind Resin and Buffer Kit , Binding Buffer 是500 M NaCl, 20 mM Tris-HCl, 5 mM imidazole, pH 7.9(加了5%的甘油稳定蛋白和0.5%的tween降低非特异结合),elution buffer是 500 M NaCl, 20 mM Tris-HCl, pH 6.0 20mM~500mM的咪唑浓度梯度,和resin结合过夜,每个浓度只洗了一次。感觉仍然结合率不高(看lysis液和unbound相比),怎么办啊??这次elution部分我上样比较多,大概每条lane相当于10ml的菌液,所以看起来量比较多。
现在看来好像50mM的咪唑就可以把我的目的蛋白洗脱下来了,但是好多好多的杂带啊!我下次是不是应该在20和50mM再摸一个浓度?漂洗的时候一般洗多少次才能最大限度把非特异洗下来而又不影响目的蛋白呢?
62253888.snap.jpg
chengjie79 (2014-1-18 16:04:23)
我的蛋白的完整性和纯度对于实验很关键,因此我在N端接了GST-tag,C端接了His-tag。考虑一次纯化不行,可以进行两步纯化来得到全长的纯度高的蛋白。请问应该先过镍柱比较好呢?还是先用Glutathione Sepharose 4B进行批量纯化呢?现在得到的蛋白洗脱液里面含高浓度的盐和咪唑,是否可以直接和Glutathione Sepharose 4B 孵育过夜来结合呢?这种beads和蛋白的结合会受哪些因素影响呢?
one (2014-1-18 16:05:12)
明显没有洗涤干净,20mm洗涤次数太少,加大洗涤体积,提高至20倍柱体积洗涤;外每一步必须要洗干净后再下一步洗涤,要不分步洗脱还有什么意义。
你的蛋白似乎his标签挂的不错,只是没有洗好。
ero11 (2014-1-18 16:05:29)
看你的lysis泳道,实在感觉不出有目的蛋白质表达来。我做过的His标签WB里,有时会有假阳性的结果。你有机会把纯化后的电泳样也做一下WB吧。或者在电泳时加上诱前诱后参照,好明确你所认定条带是否真的是目的条带。
ero11 (2014-1-18 16:19:39)
==============================================================
是不是得透析到Glutathione Sepharose 4B的上样缓冲液中啊,直接孵育肯定会有问题
standbyme (2014-1-18 16:20:24)
unbind里面还有很多是正常的,那些可能跟蛋白表达的时候折叠的方式有关,有一部分是怎样都挂不上柱的,没关系。你现在应该是没洗好,20mM 咪唑应该洗的体积大一点,起码10CV,然后每步洗两次,每次2CV。如果可以走梯度的话最好走梯度,那样目的带一半拖到后面就会比较纯。
standbyme (2014-1-18 16:28:11)
unbind里面还有很多是正常的,那些可能跟蛋白表达的时候折叠的方式有关,有一部分是怎样都挂不上柱的,没关系。你现在应该是没洗好,20mM 咪唑应该洗的体积大一点,起码10CV,然后每步洗两次,每次2CV。如果可以走梯度的话最好走梯度,那样目的带一半拖到后面就会比较纯。
greenbee (2014-1-18 16:28:38)
首先要确定是否真的表达了,然后确定你超声后目的蛋白是不是还稳定地存在,如果如图所示条带较亮的位置是你的目的蛋白的话建议调整方式如下:充分洗涤;样品中加入1-2mM的硫酸镍后延长结合时间;杂带较多有可能是非特异性吸附,你同时把盐浓度降下来,用100mM的氯化钠,binding buffer的pH跳到8.3。祝你好运。
tie8 (2014-1-18 16:29:00)
文献上不是说盐浓度越高非特异越少吗?为什么反而要把盐浓度降下来呢?
greenbee (2014-1-18 16:29:22)
盐浓度高时会增加疏水性蛋白的非特异性结合。
【求助】Ni柱纯化蛋白如何提高结合率?含图