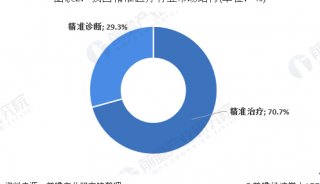
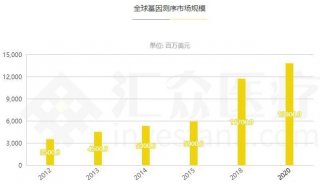
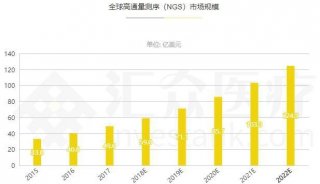
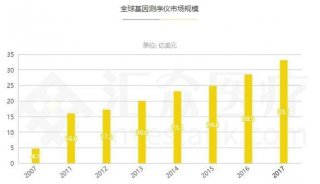
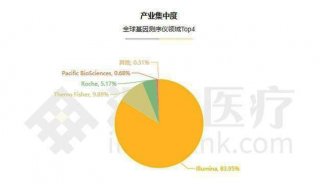
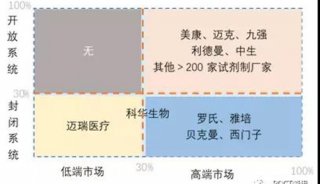
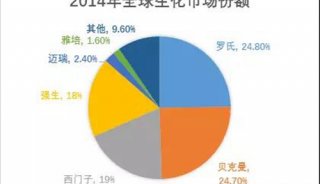
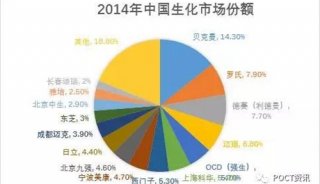
分享至
分享至



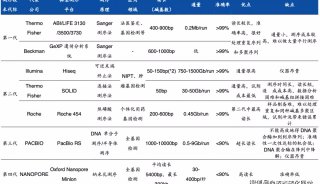
单分子测序轻松升级参考基因组
现代基因组学的最大需求之一,就是得到人类和模式生物的高质量完成基因组。目前,人们利用二代测序技术对越来越多的物种(从原核生物到真核生物)进行了基因组测序,取得了许多重要的研究成果。
不过大部分物种的基因组还中存在有大量的缺口gap,就目前的数据来说,各种已测序物种的基因组中缺口gap所占的百分比从1.3%至13%不等。这是由于二代测序技术生成的片段短,而过短的测序片段无法跨越高度重复和高GC含量的基因组区域。大量的基因组空白区域中可能存在有重要的生物学功能信息,如果无法补齐这些缺口,不仅不能获得完整的基因组物理结构图,还会给基因组信息的解读造成不小的困难。目前人们主要使用步进PCR结合Sanger测序或者illumina/454 Pair-end测序数据来填充空白区域,但是这些方法费时费力,成本高,填充效率低,无法从根本上解决问题。
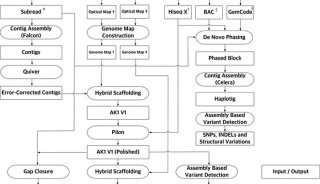
美国Baylor医学院的Richard Gibbs团队正在进行一项重要研究,利用单分子测序技术对模式生物的基因组草图进行升级。他们借助的正是Pacbio RS单分子测序的独特之处,这一技术无需PCR过程,能够轻松完成高GC含量片段和高度重复区域片段测序。该研究团队的目标是准确、自动化、快速且可重复的进行基因组升级,他们还专门开发了高度自动化的工具PBJelly,能够将Pacbio测序得到的长片段与基因组草图进行比对,填补或减少草图中的缺口,从而完善基因组草图。
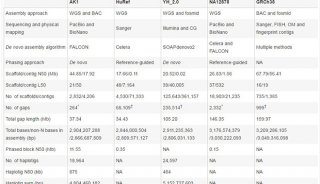
Richard Gibbs的团队利用PacBio的长读长,对两个果蝇种(Drosophila pseudoobscura和Drosophila melanogaster,基因组缺口6.7M,占基因组大小5%)、虎皮鹦鹉(M. undulates,基因组缺口155M,占基因组大小11%)、黑子白眉猴(C.atys,基因组缺口198M,占基因组大小7%)的基因组进行了升级。他们首先利用PacBio单分子测序技术进行覆盖(深度从4.4×到24×不等),然后使用PBJelly流程进行分析。研究显示长读长数据可以大大降低基因组中的缺口数,其中D. melanogaster的基因组缺口数减少了15倍,虎皮鹦鹉和白眉猴的基因组缺口数减少了1.3至2.8倍,且这些基因组的缺口大小也减少了3-6倍。随后,研究人员对随机选择的96个缺口进行了Sanger测序,验证了这一方法的可靠性。
在这一研究中,单分子测序技术帮助研究者们获得了很好的结果,大大提高了基因组的完整性,也降低了后续实验和分析的难度,远远优于传统方法。
研究人员对于含有大基因组的脊椎动物(虎皮鹦鹉基因组大小1.2G、黑子白眉猴2.8G)只进行了低深度的测序(4.4-6.8×)就能较好的改善其基因组完整性,说明PacBio测序技术在大型基因组组装领域具有强大的功能和应用潜力。
该项目还在继续进行中,研究人员将会不断的改进组装方法,提高基因组缺口的填补效率。目前,他们正在用这一方法对灵长类动物的基因组进行升级。
-
招标采购

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
焦点事件

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
焦点事件

-
焦点事件

-
市场商机

-
焦点事件

-
企业风采

-
企业风采

-
科技前沿

-
招标采购

-
焦点事件

-
科技前沿
-
技术原理
-
企业风采

-
科技前沿

-
精英视角

-
科技前沿

-
精英视角

-
企业风采

-
科技前沿

-
科技前沿

-
焦点事件

-
精英视角

-
企业风采

-
焦点事件

-
产品技术

-
焦点事件

-
焦点事件

-
技术原理

-
项目成果
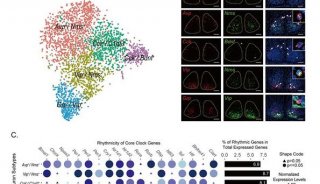
-
项目成果

-
焦点事件

-
焦点事件
-
综述
-
综述
-
综述

-
项目成果

-
焦点事件

-
项目成果
-
项目成果
-
焦点事件
-
科技前沿
-
焦点事件

-
焦点事件

-
焦点事件