
查看完整版本请点击这里:
【讨论帖】蛋白质组数据的生物信息学处理
【讨论帖】蛋白质组数据的生物信息学处理
在蛋白质组学研究中,如果使用高通量方法会得到大量蛋白质数据, 这就需要采用生物信息学的方法进行处理. 这里介绍一篇文章,希望能起到抛砖引玉的作用, 让大家讨论一下还可以用那些方法进行生物信息学处理.
在这篇论文中, 应用了合并2种检索, 非标记定量, 相对量比较(normalized and non normalized),GO term 比较, 3种算法的蛋白定位预测比较, 通路分析,蛋白修饰(包括氨基酸修饰,和蛋白降解修饰)。另外在结果表格中还列出信号肽, 跨膜区,以及是否血清蛋白分析。文章连接:
Characterization of the Vitreous Proteome in Diabetes without Diabetic Retinopathy and Diabetes with Proliferative Diabetic Retinopathy
J. Proteome Res., ASAP Article, 10.1021/pr800112g
cuturl('http://pubs.acs.org/cgi-bin/abstract.cgi/jprobs/asap/abs/pr800112g.html')
因为版权所以不能贴在这里,无法下载的可以发消息给我。
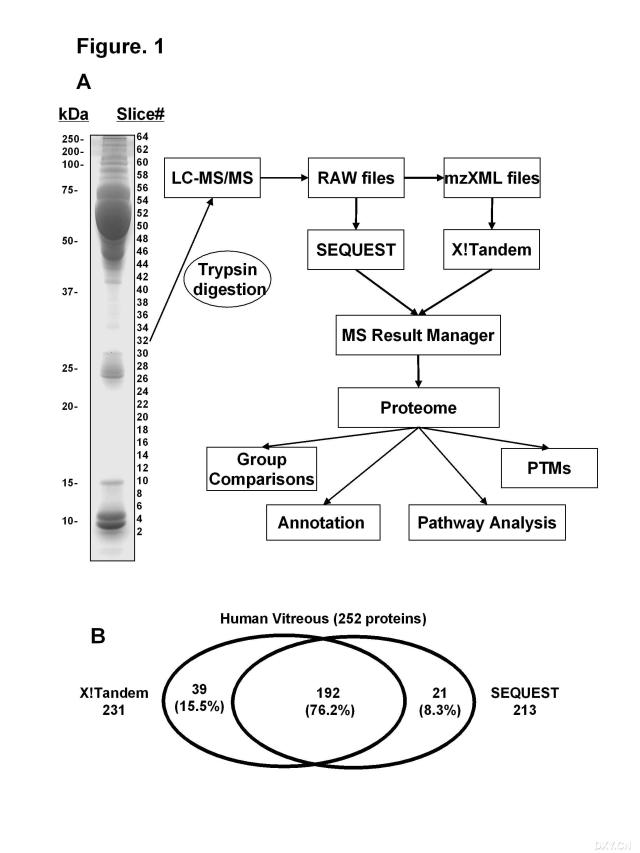
Figure 1. Proteomic analysis process and the number of proteins identified. A. Schematic of gel-LC-MS/MS analysis and data processing. B. Venn diagram of proteins identified using X!Tandem and SEQUEST algorithms. The number of proteins identified from 17 independent vitreous samples and percent of total number of proteins identified by each algorithm are shown.
查看完整版本请点击这里:
【讨论帖】蛋白质组数据的生物信息学处理
【讨论帖】蛋白质组数据的生物信息学处理
62865217.snap.jpg

最新回复
草木叉 (2013-10-10 17:40:37)
This table contains the total list of IPI, protein name, gene symbol, sequence coverage, unique peptides, spectral count of NDM, noDR, and PDR group (Mean ± SEM), p value, search engine, GO term (biological process, cellular component, molecular function), and protein subcellular localization prediction for the 252 proteins identified in this study.
54760647.snap.jpg
草木叉 (2013-10-10 17:41:04)
表格继续........
15759040.snap.jpg
草木叉 (2013-10-10 17:41:28)
Figure 2. Fractional distribution of the most abundant proteins in human vitreous. A. Chart showing a summary of the relative amounts of highly abundant proteins in PDR vitreous. B. Table showing the mean percent of number of total peptides for the 15 most abundant proteins identified in NDM, noDR, and PDR samples relative to the number of total peptides detected from respective samples.
72649213.snap.jpg
草木叉 (2013-10-10 17:41:50)
Figure 3. Comparison of proteins abundance in noDR or PDR vitreous relative to NDM vitreous. Ratio of the mean total peptides detected in noDR or PDR groups relative to the NDM group. The absence of protein detection in a group is indicated by > 20 fold.
53715041.snap.jpg
草木叉 (2013-10-10 17:42:14)
GO term, 蛋白质定位预测比较:
Figure 4. A. Frequency of Gene Ontology terms in human vitreous proteome annotation. Predicted protein subcellular localization by MultiLoc(, TargetP(C) and SubLoc(D).
10589614.snap.jpg
草木叉 (2013-10-10 17:42:41)
Ingenuity分析。 论文中以表代替图。
Proteins related to complement and coagulation cascades differently changed in human vitreous proteome in patients with NDM or PDR。(red: increase, blue: decrease)
38158743.snap.jpg
草木叉 (2013-10-10 17:43:05)
蛋白修饰分析:
Figure 5. Identification of protein fragments in the vitreous. A. Schematic of peptide coverage for proteins with a low than predicted molecular weight. The location of peptides identified is indicated. B & C. The spectral count for cadherin-2 and vitronectin on SDS-PAGE at different molecular weight from gel slices.
38618330.snap.jpg
草木叉 (2013-10-10 17:43:38)
蛋白修饰分析, 各组比较(没在论文中)
A. Frequency of protein modification in human vitreous proteome.
B. Comparison of protein modification among NDM, noDR and PDR groups.
28885520.snap.jpg
ukonptp (2013-10-10 17:44:03)
有几个问题想请教:
1. 对你的蛋白降解修饰很感兴趣, 好象没在其他文章上看过相关分析,也许是你定量分析方法的一个优势所在. 请问对绝大部分的1-D PAGE上的蛋白质, 蛋白质在不同bands中的相对定量是否有一定规律? 是否呈现正态分布? 象你文章中的vitronectin蛋白好象就没有任何规律?
2. 我感觉对数据集的生物信息学分析, 意义有限. 其他领域的科研人员能够从中间得到的信息比较有限. 其意义和出口还是要从中选择一些有意义的东西往后深入(就象你们Nature Medicine的工作), 风险很大,需要很强的背景知识. 也许这就是所谓的系统生物学研究的一部分. 不知道Dr. Gao怎么理解? 谢谢.
3. 还有对与于亚细胞器的蛋白质组数据集, 进行生物信息学分析, 该如何开展? 例如细胞膜, 进行蛋白质相互作用和pathway分析好象都受到了限制, 如纯度问题的影响.
再次感谢.
草木叉 (2013-10-10 17:44:38)
谢谢。
这个项目是我们第一个从2D Gel 转到 1D PAGE + LC/MS/MS的研究, 当时方法并不很成熟,病人样品有限, 所以监测到的蛋白总数不是太多, 现在我们使用1D GEL + 1D LC/MS/MS可以最多检测到1800个蛋白。
基于1D GEL + 1D-LC/MS/MS的蛋白组研究比较2D Gel based和2D-LC/MS/MS (Shotgun)有一些比较显著的优点。 1D GEL + 1D-LC/MS/MS 比2D Gel based 方法主要是减少很大工作量, 可以容易得到整个样品的蛋白质, 而不是数个差异点, 而且不论使用标记和非标记方法得到的定量结果都理论上比2D准确。 因为在2D胶上每个点通常很多蛋白, 一个蛋白通常分布在几个点。1D GEL + 1D-LC/MS/MS 比2D-LC/MS/MS (Shotgun)的比较也有如下优点:一是可以得到蛋白的实际分子量, 可以应用在蛋白酶切修饰的研究。 如果蛋白被修饰而出现多种分子量形式可以显示在1D gel上。 另外的一个优点是因为蛋白被分离, 所以用以低丰度的蛋白更容易被发现。但这个优点我不敢肯定,因为我们没有直接比较这两种方法。 如果有使用2D-LC/MS/MS 的实验室可以讨论一下Shotgun方法的检测灵敏度。现在我们使用1D GEL + 1D-LC/MS/MS (LTQ)可以检测到1ng左右(基于发现2个唯一多肽)。
1D GEL + 1D-LC/MS/MS的缺点是须使用nano-LC spray source而不能用MALIDI source, 2D Gel based就没有这种限制。 比shotgun 方法工作量大,LC/MS/MS时间也长很多, 我们现在的protocol需要60小时的LC/MS/MS完成一个样品的检测。
蛋白酶切修饰没有找到规律, 我们只发现几个蛋白有这种现象。 但是对于一种蛋白来讲, 每个样品都是一样的。 我们使用这种方法在另外的一个研究中发现一个很有意义的酶切现象, 结果还没有发表。
草木叉 (2013-10-10 17:44:55)
继续。
生物信息学分析是对结果分析,归纳,理解的一个方法。蛋白质组和基因组方法都是一样的,当我们发现数十种数百种有差异蛋白时,只有从这种方法入手,对富集的每个分类,每个pathway的单个蛋白进行更进一步查阅, 找出有可能有生物学意义的蛋白进一步研究。所以,生物信息学分析是一个筛选过程。
选择哪个蛋白进行深入研究是有很多风险,但部分是靠运气的。就我们的这个研究项目, 我们选择了几种蛋白进行了研究, 集中在3个蛋白上,只有CA1得到比较好的结果,另外2个都失败了。其中一个同事对一个蛋白进行了3年的研究,最后空手而归。
我不是很明白你的最后一个问题。 细胞器定位分析是根据GO annotation,另外结合计算预测。 大概80-90%的蛋白有GO注释,有细胞定位的注释的就更少, 所以需要对没有GO注释的要用计算预测的方法进行定位 (图4 B C D)。 我看过也试过不少预测工具,对分泌蛋白和细胞膜蛋白定位比较可信。 我喜欢使用CBS Prediction Servers的预测工具, 特别是signalP, TargetP, TMHMM (表中). 这些工具能批量输入, 而且最近新添加的SOAP功能可以很容易整合到自己的软件工具中。
我也试过很多pathway和GO分析工具。感觉ingenuity和DAVID比较好, ingenuity是商业软件,价格很高,可以做定量比较,显示蛋白表达的高低。 DAVID是免费的,只是对蛋白或基因进行富集分析。但对蛋白质组研究够用了。
我用的GO Slim分析是一个嵌在我的蛋白质组结果软件包的一个模块, 图4 A和表中的结果是用它分析的。
ukonptp (2013-10-10 17:45:33)
谢谢,很受用.
还是接上面的问题.
1.请问如果蛋白被修饰而出现多种分子量形式可以显示在1D gel上,多大范围以内,您认为它不是co-migration, 而是修饰,还有怎么进一步筛选修饰形式?
2.我的上面最后一个问题是想问,对于亚细胞器的蛋白质组数据集,该如何开展有针对性的生物信息学分析,定位肯定是最基本的,没问题.也许可以发现在生理或病理状态下,蛋白质定位方面的改变.但是象分析ppi和pathway时就有问题,因为不知道鉴定的蛋白质是否是该亚细胞器真正定位的蛋白质?除了实验样品的纯度提高以外,生物信息学是否能够做点什么?
谢谢.
草木叉 (2013-10-11 10:41:14)
1.请问如果蛋白被修饰而出现多种分子量形式可以显示在1D gel上,多大范围以内,您认为它不是co-migration, 而是修饰,还有怎么进一步筛选修饰形式?
不考虑范围, 看蛋白分布是不是出现2个或多个峰。
2。生物信息分析不能帮助分析病理改变引起的蛋白定位改变。
zsxan1990 (2013-10-11 10:41:36)
你好:楼主。
我是生物信息学的初学者,原来是学计算机的,现在硕士专业是生物,想做“用生物信息学方法分析蛋白方面”的课题,请问要从哪方面着手啊,非常感谢!
草木叉 (2013-10-11 10:42:58)
蛋白质研究很多都需要生物信息处理,比如:
蛋白结构分析:蛋白质序列分析,像motif, pattern识别,蛋白定位, 跨膜结构,序列同源性分析等, 二级三级结构预测。
蛋白功能分析:蛋白功能,pathway分析,相互作用分析,数据库建立等。
蛋白质质谱分析相关:质谱信号预处理,标记和非标记算法,蛋白质组结果处理。
其实每个功能都有很多论文和软件可以查阅和使用,但并不代表你不能去接着做。我有个体会,计算机科班出身做出的生物信息软件有时不会考虑到如何方便做基础研究人员的应用, 网上可以使用的GO分类的软件有数十种, 但就是找不出一个可以按照基础研究人员思维使用的软件,所以做生物信息软件一定要和作基础的合作。我做的软件是关于蛋白质非标记定量分析,蛋白质组结果处理,蛋白功能分类。如果你做与这些相关可以交流,别的蛋白分析我不懂。
yes4 (2013-10-11 10:43:37)
多谢楼主您给予的指导和建议,对我很有用。
我想做的也是和你的差不多,对蛋白功能和蛋白质组结果处理比较感兴趣,具体的现在还没定下来,正在寻找方向。
可能以后的学习过程中,还要请教您!
再次感谢您!
yes4 (2013-10-11 10:44:09)
楼主对iTRAQ的数据分析有何意见和建议?
草木叉 (2013-10-11 10:44:49)
我们的质谱仪不能做iTRAQ试验, 所以没有这方面的经验. 在刚出现这个技术时, 感觉理论上很好, 所以看了一些介绍.这个方法结合shotgun方法应该是很好的.
现在有不少配套的iTRAQ定量分析软件, 如果要自己写程序处理, 应该比lable free和iCAT简单,原因:
1. 定量分析是在MS2水平的.
2.不需要打开含有MS1数据的文件 (提交检索的数据都还有MS2 ion intensity 数据)
3.在114-117 (或114-121 8plex)这个范围, 数据干扰应该少
4.如果丰度高蛋白,会检测多个多肽, 这样会有很多比值, 容易定量准确.
分析流程图:
11630110.snap.jpg
jkobn (2013-10-11 10:45:14)
想问一哈,对于比较蛋白质组学结果的生物信息学处理,有时在不同的数据库会有不同的结果,这怎样选择和判断啊?
草木叉 (2013-10-11 10:46:30)
检索时最好用同一数据库, 如果不同数据库, 比较多肽序列, 绝大多数情况是一样的蛋白, 不一样的名称, ID.
也可以用gene name去统一, 不同数据库, 不同isoform的蛋白通常是一个gene name.
如果比较不同论文中的数据可以用IPI数据库中(cuturl('ftp://ftp.ebi.ac.uk/pub/databases/IPI/current/')) xrefs 交互关联表查询.
【讨论帖】蛋白质组数据的生物信息学处理