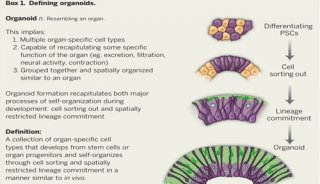
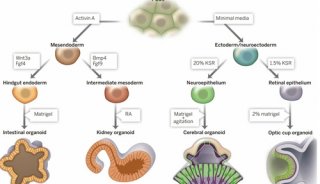
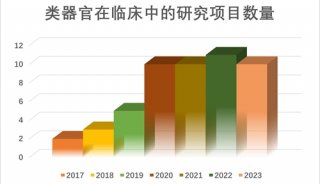
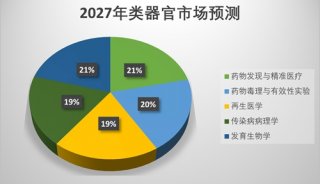
美众议院通过“21世纪治愈法案”
【新闻事件】:美国众议院近日以344比77的投票结果轻松地通过了争论已久的“21世纪治愈法案”(21st Century Cures Act)。“21世纪治愈法案”是新药开发立法的又一个里程碑,不仅进一步推动FDA对新药评审的改革,也赋予国立卫生研究院(NIH)更多的研究资源,促进基础医疗研究的发展。“21世纪治愈法案”在2014年4月由众议院能源委员会提出,其实施将改变国立卫生研究院(NIH)多年来一直停留在90亿美元预算的局面,在今后的5年里将增加87.5亿美元的财政拨款,同时FDA的经费也要增加5.5亿美元。
【药源解析】:“21世纪治愈法案”由共和党众议员Fred Upton和民主党参议员Diana DeGette联合提出,旨在改革药品监管政策,加快治疗亟需的药品审批,弥补医学研究、医药产业与药品监管之间的缺口。众议院能源委员会在经过有患者权益组织、非盈利团体、学术界、产业界、监管机构、立法机构成员参加的12次圆桌会议和8次国会听证之后,在2015年1月公布了“21世纪治愈法案”的讨论稿并公开征询各方意见。5月21日,“21世纪治愈法案”的修订稿获得众议院能源委员会举手投票通过。今天通过的“21世纪治愈法案”主要包括两个目标,一是进一步改革药品监管流程,尤其缩短那些治疗亟需药品的审批;另一个是加强基础医疗研究,鼓励医学创新。
众所周知,一个新药要想获批上市必须在临床上验证其疗效和安全性,而且这些临床实验通常是含有足够样本的,多中心、随机、双盲、和对照实验。美国FDA是全球药品监管机构的标杆,而且在过去十年FDA的成绩有目共睹,推出了快速通道、优先评审、加速审批、以及“突破性药物”等多种渠道缩短尤其是未满足市场需求药品的审批时间,曾创造了从收到申报到批准上市只花了4天的神话。“21世纪治愈法案”要求FDA进一步整合药品监管流程,强调患者在药品审批中的作用。赋予药品审批更多弹性,比如对抗生素的评审可以采纳小型临床实验结合临床前结果,接受生物标记或其它替代临床终点(surrogate endpoints)。
笔者以为加速药品的审批流程不能以降低药品疗效标准和损害患者健康为代价。而且正如今天药源讨论的那样,我们还没有进入“共产主义”,没有达到社会资源的“无限丰富”。人类社会有限的资源应该鼓励那些和现有标准疗法有明显区分的新药开发活动。事实上这也是部分业界专家对“21世纪治愈法案”的担忧。新法案是一把双刃剑,一方面“shorter or smaller clinical trials”或采用替代临床终点能明显加速新药开发的产出,另一方面势必在不同程度上影响新上市药品的质量,同时也会降低尤其是那些现行治疗金标—这些经过大型临床实验验证后上市药品的价值。而且“evidence from clinical experience” including “observational studies, registries, and therapeutic use”用作上市药物新适应症的申报,在极大程度上鼓励了未经临床验证的“处方外使用”。这些上市后统计结果虽然能提供一些疗效和安全性数据,但远不如那些随机的对照实验数据来得可靠。事实上,FDA对大约三分之一新药的审批只使用了一个平均有760试用者参与的关键临床实验结果。三分之二临床实验的真正实验周期不超过半年,包括哪些治疗终身使用的治疗药物。而且FDA对绝大多数药物的审批时间不超过10个月,所以说FDA并没有成为新药开发的瓶颈。
虽然“21世纪治愈法案”需要获得参议院通过和总统签署才能生效,但即使最终获得实施也必须警惕被“滥用”,加速新药开发不能以降低审批标准为代价,加强基础医疗研究,提高新药开发的效率才是关键。