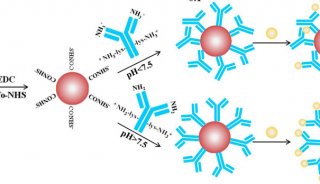
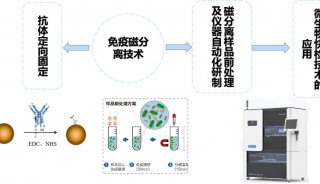
宏基因组学:查明微生物身份新手段

背景
《美国医学会杂志》(JAMA)近期刊发了一篇题为《利用不需培养的宏基因组学测序技术研究产志贺毒素大肠杆菌O104∶H4暴发株》的研究论文,引起了学界和社会的广泛关注。这篇论文中应用了一种称为“宏基因组学”的技术方法,不通过培养,直接从病人样品中检测分析其携带的病原微生物,甚至可以发现用常规方法难以检出的病原菌。由此,大家都开始关注一个共同的话题,是不是已经找到了一个能够解决突发传染病疫情病原鉴定的金刚钻呢?是不是我们以后再也不会像面对当年SARS疫情那样手足无措呢?
病原检测技术的尴尬
众所周知,人类肌体发生感染,其根本原因都是被病原微生物侵入了人体器官或组织,直接或者间接造成对人体的伤害。世界上到底有多少种微生物,目前没有人能够明确这个数字。
人类对于微生物的认识与研究主要是建立在纯培养基础上,然而研究发现通过纯培养方法估计的环境微生物多样性不及总量的1%。对于临床常规的细菌鉴定,如金黄色葡萄球菌、大肠埃希菌、鲍曼不动杆菌和铜绿假单胞菌等,传统的实验室培养方法是可靠的,但传统的培养方法也有一定局限性。比如,有相当比例的病原菌是不可以培养的;再如,在2011年德国产志贺毒素大肠杆菌(STEC)O104∶H4暴发感染时,由于这是一个既往没有报道过的新的亚型,没有相应的诊断试剂可供进行临床检验,这就使得该菌的鉴定非常困难。
现有的常规检测手段只能针对已知的病原微生物,对于未知病原,只能依靠一个长期、烦琐、严格的流程来进行判定。
随着基因组学的发展,科学家发现,绝大多数微生物都以核酸(DNA或RNA)作为遗传物质,而且微生物的基因也具有其明确的种属特征,因此对核酸进行检测分析逐渐成为了微生物检测的金标准。在这一基础上,科研人员开始尝试利用宏基组学来检测微生物。
宏基因组学的潜力
宏基因组学(metagenomics)最早是在1998年由 Handelsman 等提出的,意思是指生境中全部微生物基因组的总和。宏基因组学研究的工作流程一般可以概括成,样品采集、核酸提取、大规模测序、数据比对检索分析、生物学功能分析等。
整个工作流程中,并没有对真正的研究对象(目标微生物)进行分离、纯化和培养富集,换言之,这一研究手段并不在乎样品中有何种微生物,也不在乎有多少,而是只要在样品中,就统统把核酸序列测定出来,然后再通过和已有的核酸序列数据进行比对,判断已知种类和对未知种类进行预测。
这样的研究方法摒弃了此前对微生物的分析手段中对于可培养性的依赖。 利用这一研究手段,可以不用采取成功率和效率低下的培养方法,就能获知病原的种类,对于感染性疾病的预防、控制、诊疗和预后都具有莫大的潜力。
宏基因组学的瓶颈
几乎任何感染性疾病的诊疗,最关键的环节之一是明确病因。常规的检测技术包括定量PCR技术、基因芯片技术、酶联免疫技术、细菌生化检测技术,以及细菌培养技术等。这些技术面临着检测目标单一、耗时长、操作复杂、需多种技术相互印证、只能针对已知目标等缺陷,当遇到危急状况或暴发性新突发疫情时,现有技术手段就很难满足时效性和精确性的要求。
宏基因组学技术则能够满足这样的需求,应用这一技术可以一次性发现所有病原的存在状况,同时直接通过核酸序列进行判别,具有其他技术不可比拟的准确性优势。如果能够将这样的诊断手段应用于临床实践,将极大提高对病原微生物的判明效率和准确率,以利于具有针对性的指导治疗。
然而,这一技术的应用还面临着灵敏度、检测速度、操作性和经济性的瓶颈。宏基因组学研究中最麻烦的一点就是干扰数据的比例过高问题,80%~90%的数据都是相对无用的,相当于大海捞针,需要从海量数据中挖掘少得可怜的有效数据。同时,整个分析过程包括了样品处理、核酸提取、建测序文库、高通量测序、数据分析等环节,检测速度和操作性目前还无法向基层应用进行普及,高通量测序的成本虽然已大幅降低,但仍然难以进入普通医疗市场。
利用不需培养的宏基因组学测序技术研究产志贺毒素大肠杆菌O104∶H4暴发株
病菌菌株的鉴别对于流行性疾病暴发的控制至关重要。然而,传统的基于细菌培养的诊断方式却困难重重,特别是当不存在爆发毒株的特异性诊断检验方式时更是如此。宏基因组学方法从多种微生物混杂样品中提取DNA直接用于测序,被认为是一种不需要实验室培养即可完成爆发菌株鉴定的无限制临床检验平台。为了探索宏基因组学的潜在价值,英国伯明翰大学微生物感染研究所的Loman NJ博士等人进行了深入研究,他们发现在某一腹泻性疾病爆发时,宏基因组学具有作为无限制的、独立于培养的、用来识别和描述细菌性病原体特征方法的潜力。所面临的挑战包括加速及简化工作流程、降低成本及提高诊断的敏感性,而所有这些可能转而依赖于基因测序技术的改善。相关论文发表于国际权威杂志JAMA 2013年4月最新一期在线版。
在该项回顾性研究中,研究者选择了2011年德国产志贺毒素大肠杆菌(STEC) O104:H4毒株流行时的45例腹泻患者粪便样本并进行了分析。样本经3期分析后送交进行高通量测序(2012年8月至9月)。1期分析中,研究者利用从头测序方法得到了流行菌株的基因组草图。2期分析中,研究人员鉴别了每个样本中爆发菌株基因组覆盖的深度。3期分析中,每个样本的测序结果都与其他未知菌株作比较,以鉴定出那些属于非流行菌株的病原菌。主要结局为以基因组测序数据的覆盖率进行粪便样本中流行菌株的鉴定和特征描记。
结果显示,在1期分析中,得到了STEC流行菌株的基因组草图。2期分析中,研究人员以大于10倍的覆盖范围从10个样本中找到了该爆发菌株的基因组,并在26个样本中以大于1倍的覆盖范围找到了该爆发菌株的基因组。研究人员在40例STEC-阳性的样品中发现有27例(67%)含有来自志贺毒素基因的序列。在3期分析中,研究人员找到了难辨梭状芽孢杆菌、空肠弯曲菌、简明弯曲菌及肠道沙门氏菌的基因序列。
该研究用宏基因组学(对从复杂的微生物样品中提取的DNA进行直接测序)对2011年德国产志贺毒素型大肠杆菌爆发所做的分析中,研究人员成功地对一个疾病爆发菌株的基因组序列进行了重建;这些结果表明用这种方法具有比基于细菌培养的方法更快识别并描述细菌性病原体特征的潜力。
研究人员最终得出结论,在某一腹泻性疾病爆发时,宏基因组学具有作为无限制的、独立于培养的用来识别和描述细菌性病原体特征方法的潜力。所面临的挑战包括加速及简化工作流程、降低成本及提高诊断的敏感性,而所有这些可能转而依赖于基因测序技术的改善。