







大连化物所生物分子模拟理论方法研究取得系列进展
随着生物大分子实验技术的飞速发展,越来越巨大和复杂的分子体系被发现和鉴定出来,凸显了生物分子体系本身特有的多尺度特性,而分子动力学模拟作为生物分子功能解析的强有力工具和研究手段,已经必不可少。但是由于这些体系包含的原子数目巨大,从几千到上百万,行使功能所涉及到的时间尺度从皮秒到毫秒,解析他们行使功能的分子机制和微观机理非常困难。
现有的全原子分子模拟研究手段和方法还无法对这样的体系进行高精度和长时间动力学模拟。自2009年以来,中国科学院大连化学物理研究所分子模拟与设计研究组着眼于上述问题,针对生物体系自身独有的多尺度特性,开展了多精度分子模型的建立和动力学模拟方法发展。
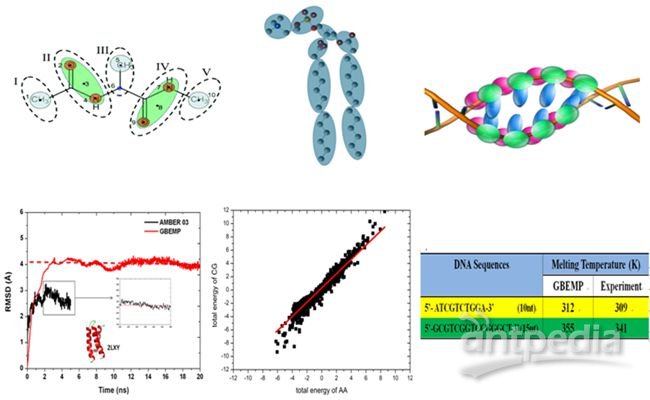
通过巧妙结合全原子可极化分子力场的高精度与粗粒化和刚体动力学模拟的计算速度快两大优势,该研究组创新性地提出和发展了可描述非球形粒子之间广义范德华相互作用的Gay-Berne势、可高精度描述粒子之间静电相互作用的多极距展开势EMP、新型高精度粗粒化分子模型GBEMP;针对组成蛋白质的20种标准氨基酸、DNA、RNA和细胞膜磷脂等一系列生物分子体系都进行了详细的理论模型构建以及参数化;利用这一高精度粗粒化分子模型的生物分子动力学模拟在精度上与全原子模型高度一致,同时,计算速度却快10-100倍,实现了精度与速度的优化匹配。相应的系列文章分别发表于J.Chem.Phys. 2011,J. Mol. Model. 2012,J. Chem. Theory Comp. 2014,J. Comp. Chem. 2015,J. Chem. Theory Comp.2016。
针对高精度理论模型计算速度耗时这一生物分子模拟关键理论问题,该研究组提出和发展了增强型采样算法与计算机图形芯片GPU相结合的软硬件加速计算方案,在国际上率先实现了高精度、高效率生物分子模拟计算程序,比之前的计算速度提高了5至10倍。作为中国大陆地区唯一的受邀者于2015年7月在美国Snowmass召开的生物分子模拟与自由能计算国际会议上作邀请报告,文章近期以封面形式发表在J. Comp. Chem 2016。
大连化物所生物分子模拟理论方法研究取得系列进展
