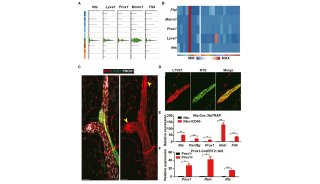
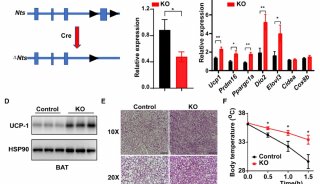
袁钧英发Cell综述 深度解析细胞坏死
来自哈佛医学院华人女科学家袁钧英教授,与另外一位学者发表了题为“SnapShot: Necroptosis”的综述性文章,重点论述了由RIPK1和RIPK3激酶介导的细胞坏死,指出抑制细胞坏死将能用于人类疾病的治疗。相关文章公布在7月11日Cell杂志上。
袁钧英教授是著名的华裔科学家,与施一公,饶毅等齐名。她一直从事细胞死亡的相关研究,2005年发现了一种细胞非调亡性的程序性死亡,英文称为 necroptosis,是国际权威之一。她还获得了美国卫生研究所(NIH)颁发了的NIH主任先驱奖(Director\'s Pioneer Award)。2000年起任哈佛大学医学院终生教授,2004年被复旦大学国家重点实验室管理委员会批准为遗传工程国家重点实验室海外客座教授。
细胞的死亡机制一直是生物医学研究的核心热点之一,目前公认的主要细胞死亡类型有三种,第一种是“坏死”,第二种是“凋亡”,第三种是“自噬”。凋亡和自噬均需要能量和合成新的蛋白质,是一个细胞自我调控的主动过程,因此也被称为“程序性死亡”。
其中细胞坏死(Necroptosis)最初被认为是一类因病理而产生的被动死亡,这些细胞的膜通透性增高,致使细胞肿胀,细胞器变形或肿大,最后细胞破裂。因此被认为是无序的过程,无从进行调控,但是RIP1和RIP3调控研究改变了这种误解,在2005年,这种与死亡受体配基相关的程序性坏死被命名为 “necroptosis”。
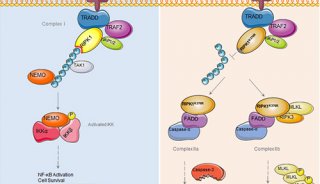
在这篇综述中,两位学者指出,这种由RIP1和RIP3介导的调控性细胞坏死具有一些独特的细胞特征,将其与细胞凋亡区分开来。细胞坏死是由细胞外刺激激活的,这些刺激经研究发现,还能用于激活炎症,细胞死亡。细胞坏死的细胞内信号途径包括坏死信号复合体 necrosome,MLKL激活。研究表明,抑制细胞坏死能减轻多个小鼠模型的病理情况,这为治疗人类疾病提出了新的希望。
之前的研究表明,RIPK1参与了依赖或不依赖RIPK-3的信号通路,这些信号通路决定了细胞的死亡及炎症反应。遗传敲除ripk1基因会引发出生后致死,但是缺失RIPK1,RIPK3 ,或者caspase -8,FADD蛋白的动物却能存活下来,甚至生长至成熟个体。
而最新的一项研究表明RIPK1能阻断Caspase-8 和RIPK3介导的出生后早期死亡。体外实验表明RIPK1能限制caspase-8依赖性,TNFR诱导的细胞凋亡,但缺失RIPK1, RIPK3和TNFR1的动物能存活至成年,研究人员还发现RIPK3能增加ripk1−/−小鼠的致死率,这说明前者的活性受到了RIPK1的抑制。
而TNFR诱导的RIPK3依赖性细胞坏死需要RIPK1,如果细胞缺少RIPK1,就更易发生由poly:C或干扰素引发的细胞坏死。 这些研究结果揭示了RIPK1在出生后小鼠中的复杂作用,也为进一步了解RIPK1相关的FADD-caspase-8,RIPK3-MLKL信号通路调控提供了新的数据资料。
此外,清华大学的施一公研究组去年报道了小鼠RIP3激酶结构域, MLKL激酶样结构域,以及两者之间形成的二元复合物的晶体结构。从中研究人员发现,RIP3和MLKL都具有典型的激酶折叠,活性构象中存在自由的 RIP3,而MLKL-RIP3的结合会通过AMP-PNP形成非活性构象,得以稳定。在RIP3-MLKL复合体中,RIP3会发生αC螺旋和活性环的显著构象改变,而MLKL相应结构元件也会发生变化。
研究人员还发现,RIP3介导的MLKL的磷酸化,是一种重要的下游信号,对于RIP3和MLKL之间形成稳定的复合物结构也是必不可少的。这些研究成果为搭建RIP3介导的细胞坏死信号途径框架提出了新分子机制,将有助于从结构生物学方面了解RIP3细胞坏死作用机制。
-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件
-
科技前沿

-
焦点事件

-
科技前沿

-
焦点事件

-
项目成果

-
科技前沿

-
焦点事件
-
科技前沿

-
技术原理

-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件
-
焦点事件

-
焦点事件

-
项目成果
-
项目成果