北京生科院提出微生物组解析新技术
1月23日,国际学术期刊《自然-通讯》(Nature Communications)在线发表了中国科学院北京生命科学研究院计算基因组学实验室赵方庆团队题为MetaSort untangles metagenome assembly by reducing microbial community complexity 的研究成果。该研究首次提出基于降低物种复杂度策略的复杂微生物组结构解析的新技术。
微生物组群落结构的多样性是群落发挥生态功能的重要基础,因此解析复杂微生物群落结构一直是宏基因组研究的重点但也是难点。以往研究中对微生物群落结构的解析主要是通过与参考数据库比对来实现的,所以未知环境下微生物群落的研究受到极大限制。从单个细胞水平获取微生物基因组来看,单细胞测序技术可以在复杂微生物群落的基因组结构解析方面有着重要的应用潜力。然而,由于微生物单细胞测序技术具有高成本、低成功率、所产生的数据覆盖度高度不均一等固有缺陷,使得它在微生物组学研究中的应用受到很大限制。
为了解决上述问题,赵方庆团队提出了基于降低物种复杂度策略的微生物组结构解析的新技术——metaSort,它将单细胞测序和全基因组随机测序技术相结合,以获取微生物群落中不同物种基因组的完整序列。metaSort利用流式细胞术对宏基因组样品中的细菌进行排序,然后分选出指定区间内指定数目的细菌子集。随后,利用单细胞技术对每个细菌子集进行扩增测序。为了利用原始的宏基因组和分选的细菌子集信息,他们还提出了两个新的算法模型:BAF和MGA。这两个方法可以利用子集中富集细菌的部分基因组序列,从原始宏基因组数据中回收目标基因组序列,并对这些序列进行拓扑组装和变异识别。研究中将该技术应用到口腔和肠道微生物样品中,均证明该方法的有效性。他们又进一步对未知微生物群落——海藻表面共生微生物进行了研究:仅通过3次流式细胞分选,就成功获得72个接近完整的微生物全基因组序列。通过三代测序技术对拼接后的基因组序列进行验证,表明metaSort方法具有很高的准确性。metaSort已公开发布在免费的开源网站SourceForge上,以方便相关研究者下载使用。
metaSort方法的优势在于为用户提供了灵活的方式获取新环境样品中微生物的基因组序列,用户可以自行控制是分选单个细胞还是范围和数目较大的细胞组成,并且可以控制分选细胞的数量和区域较小宏基因组的复杂度。与传统的单细胞测序相比,metaSort分选出的细胞子集交集很小,意味着通量的提高和成本的降低。此外,其他的分选方式,例如特异性的核酸探针和抗体标记的磁珠都可以应用到metaSort中以获取目标细菌,这些方法都会极大地提高metaSort的应用范围,进而推动对未知环境中微生物组成、基因功能和代谢网络的研究进展。
该工作由赵方庆团队的助理研究员冀培丰和博士后张延明共同完成,并得到国家自然科学基金和科技部重点研发计划的资助。
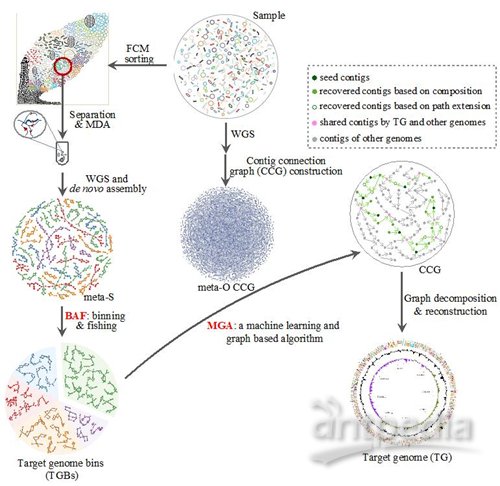
metaSort技术流程
-
焦点事件

-
焦点事件

-
科技前沿

-
焦点事件

-
项目成果

-
项目成果
-
焦点事件