


简介

锂系电池分为锂电池和锂离子电池。锂离子电池广泛应用于汽车、手机和笔记本电脑等设备,通常人们俗称其为锂电池。电池一般采用含有锂元素的材料作为电极,是现代高性能电池的代表。而真正的锂电池由于危险性大,很少应用于日常电子产品。目前,使用越来越多,发展更快的是锂离子电池。成为大多数电子产品和动力电池的关键能源供给装置。锂离子电池重量轻,锂离子电池不含有金属态的锂,是可以充电的,能量密度大,循环使用寿命长,且不会对环境造成污染。在锂离子电池正极材料合成的产业化进程中,对高含量的锂进行快速、准确的控制分析和样品系统分析具有重要意义。
石油焦炭和石墨作负极材料无毒,且资源充足,锂离子嵌入碳中,克服了锂的高活性,解决了传统锂电池存在的安全问题,正极LixCoO2在充、放电性能和寿命上均能达到较高水平,使成本降低,总之锂离子电池的综合性能提高了。预计21世纪锂离子电池将会占有很大的市场。
锂离子二次电池充、放电时的反应式为:

发展过程
1970年,埃克森的M.S.Whittingham采用硫化钛作为正极材料,金属锂作为负极材料,制成首个锂电池。锂电池的正极材料是二氧化锰或氯化亚砜,负极是锂。电池组装完成后电池即有电压,不需充电。锂离子电池(Li-ion Batteries)是锂电池发展而来。举例来讲,以前照相机里用的扣式电池就属于锂电池。这种电池也可以充电,但循环性能不好,在充放电循环过程中容易形成锂结晶,造成电池内部短路,所以一般情况下这种电池是禁止充电的。
1982年伊利诺伊理工大学(the Illinois Institute of Technology)的R.R.Agarwal和J.R.Selman发现锂离子具有嵌入石墨的特性,此过程是快速的,并且可逆。与此同时,采用金属锂制成的锂电池,其安全隐患备受关注,因此人们尝试利用锂离子嵌入石墨的特性制作充电电池。首个可用的锂离子石墨电极由贝尔实验室试制成功。
1983年M.Thackeray、J.Goodenough等人发现锰尖晶石是优良的正极材料,具有低价、稳定和优良的导电、导锂性能。其分解温度高,且氧化性远低于钴酸锂,即使出现短路、过充电,也能够避免了燃烧、爆炸的危险。
1989年,A.Manthiram和J.Goodenough发现采用聚合阴离子的正极将产生更高的电压。
1992年,日本索尼公司发明了以炭材料为负极,以含锂的化合物作正极的锂电池,在充放电过程中,没有金属锂存在,只有锂离子,这就是锂离子电池。随后,锂离子电池革新了消费电子产品的面貌。此类以钴酸锂作为正极材料的电池,是便携电子器件的主要电源。
1996年,Padhi和Goodenough发现具有橄榄石结构的磷酸盐,如磷酸铁锂(LiFePO4),比传统的正极材料更具安全性,尤其耐高温,耐过充电性能远超过传统锂离子电池材料。
纵观电池发展的历史,可以看出当前世界电池工业发展的三个特点,一是绿色环保电池迅猛发展,包括锂离子蓄电池、氢镍电池等;二是一次电池向蓄电池转化,这符合可持续发展战略;三是电池进一步向小、轻、薄方向发展。在商品化的可充电池中,锂离子电池的比能量最高,特别是聚合物锂离子电池,可以实现可充电池的薄形化。正因为锂离子电池的体积比能量和质量比能量高,可充且无污染,具备当前电池工业发展的三大特点,因此在发达国家中有较快的增长。电信、信息市场的发展,特别是移动电话和笔记本电脑的大量使用,给锂离子电池带来了市场机遇。而锂离子电池中的聚合物锂离子电池以其在安全性的独特优势,将逐步取代液体电解质锂离子电池,而成为锂离子电池的主流。聚合物锂离子电池被誉为“21世纪的电池”,将开辟蓄电池的新时代,发展前景十分乐观。
2019年10月9日,瑞典皇家科学院宣布,将2019年诺贝尔化学奖授予约翰·古迪纳夫、斯坦利·惠廷厄姆和吉野彰,以表彰他们在锂离子电池研发领域作出的贡献。
2022年二季度,锂离子电池、电子元器件、集成电路等小类行业增加值增速均在20%以上。


产品优点
1)电压高
单体电池的工作电压高达3.7-3.8V(磷酸铁锂的是3.2V),是Ni-Cd、Ni-MH电池的3倍。
2)比能量大
能达到的实际比能量为555Wh/kg左右,即材料能达到150mAh/g以上的比容量(3-4倍于Ni-Cd,2-3倍于Ni-MH),已接近于其理论值的约88%。
3)循环寿命长
一般均可达到500次以上,甚至1000次以上,磷酸铁锂的可以达到8000次。对于小电流放电的电器,电池的使用期限,将倍增电器的竞争力。
4)安全性能好
无公害,无记忆效应。作为Li-ion前身的锂电池,因金属锂易形成枝晶发生短路,缩减了其应用领域:Li-ion中不含镉、铅、汞等对环境有污染的元素;部分工艺(如烧结式)的Ni-Cd电池存在的一大弊病为“记忆效应”,严重束缚电池的使用,但Li-ion根本不存在这方面的问题。
5)自放电小
室温下充满电的Li-ion储存1个月后的自放电率为2%左右,大大低于Ni-Cd的25-30%,Ni-MH的30-35%。
6) 快速充电
1C充电30分钟容量可以达到标称容量的80%以上,磷铁电池可以达到10分钟充电到标称容量的90%。
7) 工作温度
工作温度为-25~45°C,随着电解液和正极的改进,期望能扩宽到-40~70°C。
产品缺点
1)衰老
与其它充电电池不同,锂离子电池的容量会缓慢衰退,与使用次数有关,也与温度有关。这种衰退的现象可以用容量减小表示,也可以用内阻升高表示。
因为与温度有关,所以在工作电流高的电子产品更容易体现。用钛酸锂取代石墨似乎可以延长寿命。储存温度与容量永久损失速度的关系:
充电电量 | 储存温度0℃ | 储存温度25℃ | 储存温度40℃ | 储存温度60℃ |
40%~60% | 2%/年 | 4%/年 | 15%/年 | 25%/年 |
100% | 6%/年 | 20%/年 | 35%/年 | 80%/6月 |
2)回收率
大约有1%的出厂新品因种种原因需要回收。
3)不耐受过充
过充电时,过量嵌入的锂离子会永久固定于晶格中,无法再释放,可导致电池寿命短。
4)不耐受过放
过放电时,电极脱嵌过多锂离子,可导致晶格坍塌,从而缩短寿命。
电量不足

政策标准
随着锂电行业的发展,我国首部锂离子电池强制标准于2015年8月1号正式实施。
为加强锂离子电池行业管理,提高行业发展水平,引导产业转型升级和结构调整,推动锂离子电池产业持续健康发展,2015年12月11日,工信部发布了《锂离子电池行业规范条件》征求意见稿,明确了锂离子电池企业和产品的准入规则。
在锂离子电池产业供应链的上中游,原材料和产品的质量控制需要仪器分析方法来确定正负极材料、电解液等原材料的纯度。
锂离子电池的化学性质至关重要,因此,准确可靠地分析此类样品对于确保最终产品质量符合要求来说至关重要。这需要工作的硬件和软件功能的良好协调与平衡。
该行业中对于产品质量要求也是相当严格的,微量锂的测定常采用原子吸收光谱法。对于高含量的锂,一般采用化学方法。相对而言,化学方法操作繁琐和费时,不适合产业化中的批量分析。原子吸收法具有干扰少、简便、快速等优点,在低含量元素测定尚应用较广泛,高含量测定报道相对较少。本研究采用原子吸收法对高含量的锂进行控制分析和样品系统分析的研究具有实际意义。
该行业所需检测项目繁多,包括包装、运输、存放、抗压、抗暴、耐热、充、放电、以及正、负极原材料纯度的测试等等。
所需设备,除实验室常规设备外所需仪器:
检测项目 | 所需仪器 | 对应标准 | 备注 |
石墨类负极材料 | 激光粒度仪 | GB/T
24533-2016 | |
炭复合磷酸铁锂正极材料 | ICP-OES | GB/T 30835-2014 | |
聚烯烃隔膜 | 鼓风式恒温箱 | GB/T
36363-2018 | |
废弃物回收利用的处理方法 | 破碎分选装置 热解炉 搅拌机 压滤机 废气、废水处理装置 废渣收集设备 电位滴定仪 分光光度计 ICP-OES 原子吸收光谱仪 原子荧光光谱仪 | GB/T
33059-2016 | |
正极材料检测方法磁性异物和残余碱含量的测定 | 球磨机 ICP-OES 原子吸收光谱仪 磁环套 扫描电镜-能谱仪 真空过滤装置 磁力搅拌器 电位滴定仪 | GB/T
41704-2022 | |
铝及铝合金箔 | 原子吸收光谱仪 分光光度计 热导测氢仪 高纯度石墨坩埚 碳硫分析仪 | GB/T
33143-2022 | 中所要求的GB/T 17432 |
压延铜箔 | 原子吸收光谱仪 电热恒温干燥箱 符合要求的电解器 分光光度计 | GB/T
36146-2018 | 中所要求的GB/T 5121 |
本方案只针对原材料纯度检测Pb、Cd、Cr以及主要元素Li,本方案采用原子吸收火焰法、原子荧光法进行测试,原子吸收火焰法测试主要元素Li时需要调整燃烧头偏转角度后,对高含量锂的测定进行了系统研究,考察了测定介质、酸度、吸光度、灯电流等的影响及共存元素干扰情况,对在空气-乙炔火焰中测定锂离子电池正极材料锰酸锂中高含量锂的原子吸收方法测量的多种影响因素进行了研究。
原子荧光法根据实验环境,调整仪器设置,达到下列标准要求即可。
规范引用参考标准:GB/T 39560.5-2021、GB/T 17413.1-2010。
前处理
1 试剂
使用以下试剂:
a) 水:ISO 3696,一级水。用于所有样品溶液的制备和稀释。
b) 硫酸:
1) 硫酸:p(H2SO4)=1.84 g/mL,质量分数为 95%,痕量金属级。
注:痕量金属级是指试剂的目标元素或干扰物的浓度与被测定元素的最低浓度相比低至可被忽略。换言之,试剂的目标元素或干扰物的浓度不影响实际检测。机构需谨慎选择与之对应的试剂等级。
下同。
2) 硫酸(1 :2):取 1 体积的浓硫酸[1 b)1)]和 2 体积的水[1 a)]混合。
C) 硝酸:
1) 硝酸:p(HNO3)=1.40 g/mL,质量分数为 65%,痕量金属级。
2) 硝酸:质量分数为 10%,痕量金属级。
3) 硝酸:0.5 mol/L,痕量金属级。
4) 硝酸(1 :2):取 1 体积的浓硝酸[1 c) 1)]和 2 体积的水[1 a)]混合。
d) 盐酸:
1) 盐酸p(HCl)= 1.19 g/mL,质量分数为 37%,痕量金属级。
2) 盐酸(1 :2):取 1 体积的浓盐酸[1 d)1)]]和 2 体积的水[1 a)]混合。
3) 盐酸:质量分数为 5%,痕量金属级。
4) 盐酸:质量分数为 10%,痕量金属级。
e) 氢氟酸:p(HF)=1.18 g/mL,质量分数为 40%,痕量金属级。
f) 氟硼酸:HBF4,质量分数为 50%,痕量金属级。
g) 高氯酸:p(HClO4)= 1.67 g/mL,质量分数为 70%,痕量金属级。
h) 磷酸:p(H3PO4)= 1.69 g/mL,质量分数超过 85% ,痕量金属级。
i) 氢溴酸:p(HBr)=1.48 g/mL,质量分数为 47%~49%,痕量金属级。
j) 硼酸(H3BO3):50 mg/mL,质量分数为 5%,痕量金属级。
k) 过氧化氢:p(H2O2)=1.10 g/mL,质量分数为 30%,痕量金属级。
1) 混合酸:
1) 混合酸 1:取 2 体积盐酸[1 d)1)]、1体积硝酸[1 c)1)]和 2 体积水[1 a)]混合。
2) 混合酸 2:取 1 体积硝酸[1 c)1)]和 3 体积氢氟酸[1 e)]混合。
3) 混合酸 3:取 3 体积盐酸[1 d)1)]和 1 体积硝酸[1 c)1)]混合。
m) 氢氧化钾(KOH):痕量金属级。
n) 硼氢化钾(KBH4):痕量金属级。
0) 铁氰化钾{K3[Fe(CN)6]}:痕量金属级。
P) 氧化还原剂:将质量分数为 1.5% 的硼氢化钾和质量分数为 1%的铁氧化钾加到质量分数为0.2%氢氧化钾水溶液中。加入约 800 mL 水[1 a)]至 1 000 mL 容量瓶,然后加入 2 g 氢氧化钾[1 m)]、15 g 硼氢化钾[1 n)]、10g 铁氰化钾[1 o)]至容量瓶,搅拌溶解,加水定容[1 a)]。当天配制。
q) 还原剂:
1) 还原剂 1:将质量分数为3%的硼氢化钾加到质量分数为 0.2%的氢氧化钾水溶液中:加入约800mL水[1 a)]至 1 000 011容量瓶,然后加入 2 g 氢氧化钾[1 m)]、30 g硼氢化钾[1 n)]至容量瓶,搅拌溶解 ,加水定容[1 a)]。当天配制。
2) 还原剂 2:将质量分数 4%的硼氢化钾加到质量分数 0.8%的氢氧化钾水溶液中:加入约800 mL水[1 a)]至 1 000 mL 容量瓶,然后加入 8 g氢氧化钾[1 m)]、40 克硼氢化钾[1 n)]至容量瓶,搅拌溶解 ,加水定容[1 a),当天配制。
r) 载流:
1) 载流 1:质量分数为 1.5% 的 HCl溶液。
2) 载流 2:质量分数为1% 的 HCl溶液。
s) 硫脲[(NH2)2CS]溶液:质量分数为10%。当天配制。
t) 掩蔽剂:
1) 掩蔽剂 1:质量分数分别为 5%的草酸、5%的硫氰酸钾和 0.5%的邻菲咯啉的水溶液。加入 10 g 草酸、10 g 硫氰酸钾和 1 g 邻菲咯啉到 200 mL 水中[1 a)].在低温下加热搅拌溶解,注意避免溶液沸腾。在固体结晶析出之前溶液可正常使用。当溶液颜色变深时需弃用,应配制新的溶液。
2) 掩蔽剂 2:质量分数分别为 10%的硫脲、10%的抗坏血酸的水溶液。在 100 mL 水中溶解10 g 硫脲和 10 g 抗坏血酸。当天配制。
u) 钴溶液:50mg/L。
v) 储备溶液:
1) 1 000 mg/L 的 Pb 储备溶液。
2) 1 000 mg/L 的 Cd 储备溶液。
3) 1 000 mg/L 的 Cr 储备溶液。
4) 10 000 mg/L 的 Fe 储备溶液。
5) 10 000 mg/L 的 Cu 储备溶液。
本方法中每种试剂的毒性尚未被准确评估,然而,每一种化合物都应视为对健康有潜在危害。为此 ,宜采用任何适当的方法尽可能减少上述试剂的暴露。
样品前处理方法涉及使用强酸,强酸具有腐蚀性,可引起烧伤。在处理这些强酸时应穿戴实验室防护服、手套和护目镜。
硝酸具有毒挥发性。因此消解和向样品中添加酸的操作均应在通风橱中进行,以避免有毒气体释放的危害。
等离子体产生的废气应用有效的抽风系统排出。
使用氢氟酸时应采取特别的预防措施。如氢氟酸烧伤皮肤应用氢氟酸解毒胶(含 2.5%葡萄糖酸钙的水溶胶)进行急救处理。
本方案为AAS、AFS测试方案,至少使用分析纯试剂。
2 仪器
2.1 概述:
玻璃器皿的收集和存放是微量分析的关键。由于 Pb、Cd 和 Cr 分析技术的灵敏度高,每一个样品处理步骤都要谨慎进行。所有的取样、储存和操作器具都应不含待测物。在 10%硝酸[1 c)2)]中浸泡所有玻璃器皿,室温下保持 24 h,然后用水[1 a)]进行彻底冲洗。
2.2 仪器
应使用下列设备:
a) 分析天平:精确至 0.0001 g。
b) 耐 HF 进样系统:对进样系统部件和炬管进行耐 HF 处理。
c) 氩气:纯度超过 99.99%的气体。
d) 乙炔气:纯度超过 99.99%的气体。
e) 玻璃器皿:所有玻璃器皿在使用前应用10%硝酸[1 c)2)]清洗:
1) 长颈烧瓶 100 mL;
2) 烧杯:如 100 mL、200 mL、500 mL 等;
3) 容量瓶:如 50 mL、100 mL、200 mL、500 mL、l 000 mL 等,在满足精确度和准确度的要求下,可选择其他适当的定容装置代替容量瓶;
4) 移液管:如 1 mL、5 mL、10 mL、20 mL 等;
5) 表面皿。
f) 钳金生埸:如 50 mL、150 mL 等。
g) 瓷片 :如 50 mL、150 mL 等。
h) PTFE/PFA 设备(聚四氟乙烯(PTFE)/全氟烷氧基烷煌树脂(PFA):所有设备在使用前应用10%硝酸[1 c)2)]进行清洗:
1) 烧杯如 100 mL、200 mL、500 mL 等;
2) 烧杯盖:
3) 容量瓶:如 100 mL瓶00 mL、500 mL 等。
i) 微型移液管:如 10 μL、100 μL、200 μL、500 μL、1 000 μL等。
j) 试剂瓶:用于储备溶液和校准溶液的储存。
试剂瓶由高密度聚乙烯(PE-HD)或全氟烷氧基树脂(PFA)制成。
k) 超痕量测试时,应使用全氟烷氧基树脂(PFA)或全氟(乙烯丙烯)塑料(FEP)制成的试剂瓶。在任何一种情况下,实验操作人员应确认所选择试剂瓶的适用性。
l) 电热板或砂浴锅。
m)马弗炉:可保持在 550℃ ±25℃。
n) 本生燃烧器或类似的气体燃烧器。
0) 王水消解装置 :消解装置配有时间温度微控单元、恒温加热块 、系列容器,每组都配有回流冷凝器和吸收器皿的装置。
p) 微波消解系统:配有样品架和耐高压聚四氟乙烯/改性四氟乙烯(PTFE/TFM)或全氟烷氧基树脂/改性四氟乙烯(PFA/TFM)或基于氟碳材料的其他消解罐。
针对各个实验室使用的不同型号的微波设备 ,有许多安全操作规范。使用者需要查阅专门的设备手册、文献或咨询制造厂商,以便安全正确操作微波设备和消解罐。
q) 耐热保温板。
r) 孔径为 0.45 μm 的玻璃微纤维过滤器(硼硅酸盐玻璃)和一个合适的过滤杯。
s) 原子吸收光谱仪(AAS)。
t) 原子荧光光谱仪(AFS)。
3 试料3. 1 聚合物
使用湿法酸消解法时,称取 400 mg 已经经过研磨、碾磨或切削好的样品,精确到 0.1 mg。使用干灰化法或微波消解法时,称取 200 mg 已经经过研磨、碾磨或切削好的样品,精确到 0.1 mg。
3.2 金属
称取约 1 g 样品,精确到 0.1 mg, 放置在玻璃烧杯中,如使用 HF[1 e)]则需使用 PTFE/PFA 烧杯。使用 AFS 法,所需样品量为 0.2 g。
3.3 电子件
使用王水消解时 ,称取 2 g 经研磨的样品(最大粒径:250 mm),精确至 0.1 mg。使用微波消解法时,称取 200 mg 经研磨的样品(最大粒径:250 am),精确至 0.1 mg。
4. 分析步骤
4.1 聚合物
4.1.1干灰化法
如果样品不含卤素化合物(可从先前的筛选测试获得信息).则应执行以下步骤:
a) 称取样品放入瓷坩埚[2.2 g)]中,并置于耐热隔热板[2.2 q)]的孔上。
b) 用燃烧器[2.2 n)]慢慢加热瓷坩埚[2.2 g)J保持适当通风,避免点燃样品。
c) 当样品被分解为焦块状,逐渐加热升温直到挥发性分解产物充分排出,剩下干碳质残渣。
d) 将瓷坩埚及内容物转移到 550 ℃±25 °C的马弗炉[2.2 m)],炉门轻微打开以提供足够的空气
氧化碳质残渣。
e) 继续加热直到碳质残渣完全氧化成灰烬。
f) 从马弗炉中[2.2 m)]取出瓷坩埚[2.2 g)],冷却至室温。如使用 AFS 法测试,参见 4.1.1 h)的操作。
g) 加入 5 mL 硝酸[1 c)1)],将溶液转移到 50 mL 容量瓶[2.2 h)3 )]中,并加水[1 a)]定容,得到样品的浓缩液。根据不同仪器情况,可用水[1 a)]稀释上述溶液到适当浓度进行测定。
h) 将所得溶液转移到 100 mL 容量瓶[2.2 h)3)],并加水[1 a)]定容。取 2.5 mL 溶液至100 mL烧杯[2.2 e)2)]中。将烧杯放在电热板上[2.2 1)],低温下加热直到溶液完全干燥。用少许水[1 a)]冲洗烧杯的内壁 ,加入 1.0 mL(用于测定 Cd)或 1.5 mL(用于测定 Pb)盐酸溶液[1 d)2)],稍微加热,使烧杯中的盐溶解。将溶液冷却至室温,并将其转移到 50 mL 容量瓶[2.2 h)3)]。50 mL 容量瓶中的溶液将分别按照以下步骤进行处理:
——测定 Pb 时,加水[1 a)]至刻度并混匀。
——测定 Cd 时,如果样品不含铜、铁、锌、银等杂质,加入 1.0 mL 钻溶液[1 u)]和 5.0 mL 硫脲溶液[1 s)]到容量瓶。如果样品含有这些外来金属杂质,则用 10.0 mL 掩蔽剂2[1 t)2)]代替 5.0 mL 硫脲溶液[1 s)],加水[1 a)]定容并混匀。
如果样品含有大量的卤素化合物(可从先前的筛查试验获得信息),则应执行以下步骤:
i) 称取样品至瓷坩埚中[2.2 g)]。
j) 在瓷坩埚中加入 5 mL 15 mL 硫酸[1 b)l)],然后在电热板或砂浴锅[2.2 1)]上缓慢加热瓷坩埚[2.2 g)]及其内容物 ,直到塑料熔化并变黑。
k) 冷却后,加入 5 mL 硝酸[1 c)l)]继续加热直到塑料完全被消解并生成白色烟雾。
l) 冷却后,将坩埚[2.2 g)]放置在 550 ℃±25 ℃的马弗炉[2.2 m)]中,将样品蒸发、干燥和灰化,直到碳完全燃烧。
m) 灰化结束后,加入 5 mL 硝酸[1 c)1)],并将溶液转移到 50 mL容量瓶[2.2 e)3)]中,加水[1 a)]定容。得到样品的浓缩液。根据不同仪器情况,可用水[1 a)]稀释上述溶液到适当浓度进行测定。
n) 任何样品残留物均应用离心机或过滤器分离。残留物应通过适当的方法检查(例如 XRF、碱熔法 、其他酸溶法等),确认没有目标元素损失。
4.1.2微波消解步骤如下:
——将称好的样品放入微波消解罐中,加入 5 mL 硝酸[1 c)1)]和 1 mL HF[1 e)],再加入少量(如:0.1 mL~1 mL)过氧化氢[1 k)],以促进有机物质的完全氧化。盖上消解罐盖,放在微波消解装置[2.2 p)]中。样品按照预先设定的程序进行消解 ,加入硼酸[1 j)]使氟化物络合,以保护石英等离子体炬管(适用于不具备耐酸进样系统的仪器)。冷却后,将溶液转移到 50 mL PTFE/PFA 容量瓶[2.2 h)3)]中,加水[1 a)]定容。得到样品的浓缩液。根据不同仪器情况,可用水[1 a)]稀释上述溶液到适当浓度进行测定。
注:本法不适用于 AFS 法。
4.2 金属
样品消解步骤如下:
——测定 Pb 时 ,将 4.0 mL 掩蔽剂 1[1 t)l)]加入容量瓶中,加水[1 a)]至刻度并混匀。静置约 30 min,然后用慢速滤纸直接过滤。滤液进行测试。
——测定 Cd 时 ,将 1.0 mL 钻溶液[1 u)]和 5.0 mL 掩蔽剂 2[1 t)2)]加入容量瓶中 ,加水[1 a)]至刻度。放置约 30 min 后对溶液进行测试。
4.3 电子件
王水消解步骤如下:
称取约 2 g 研磨的样品(最大粒径 250 μm)放入到反应容器中,精确至 0.1 mg,加入 30 mL 混合酸3[1 1)3)]。反应器需配备一个回流冷凝器和一个吸收皿,吸收皿中装有 10 mL 0.5 mol/L HNO3 [1 c)2)。消解时应控制温度,在室温下消解 12 h,在 120℃ 下消解 2 h。冷却至室温后,将吸收皿中的提取物转入反应容器中,溶液用 0.45 μm 的玻璃微纤维过滤器[2.2 r)]上过滤 ,用15 mL 5% HCl[1 d)3)]洗涤固体残留物四次。将溶液转移至 250 mL 容量瓶[2.2 e)3)],以 5%HCl[1 d)3)]定容,用于 ICP-OES、ICP-MS 和 AAS 检测,或转移溶液至 1 000 mL 容量瓶[2.2 e)3)],并以 5%HCl[1 d)3)]定容,用于 AFS 检测。所得溶液是样品的浓缩液。用 5% HC1[1 d)3)]将样品溶液稀释至不同测试仪器适合的浓度水平。
对于 AFS 方法,吸取 2.50 mL 样品浓溶液 100 mL 烧杯[2.2 e)2)]中,将烧杯放在电热板上[2.2 1)],低温加热直到溶液完全干燥。用少许水[1 a)]冲洗烧杯的内壁 ,加入 1.0 mL(用于测定 Cd)或 1.5 mL(用于测定 Pb)盐酸溶液[1 d)2)]。稍微加热,使烧杯中的盐溶解。将溶液冷却至室温 ,然后将其转移到 50 mL 容量瓶[2.2 e)3)]中。50 mL 容量瓶中的溶液分别按以下步骤处理:
——测定 Pb 时 ,力。4.0 mL 掩蔽剂 1[1 t)1)]至容量瓶中 ,用水[1 a)]定容混匀,放置约30 min,用 0.45 ptm 玻璃微纤维过滤器[2.2 r)]过滤。滤液进行检测。
——测定 Cd 时 ,加 1.0 mL 钴溶液[1 u)]和 5.0 mL 掩蔽剂 2[1 t)2)]至容量瓶中,用水[1 a)]定容。放置约 30 min,溶液待测。
如果过滤器上有样品残留物 ,则 应通过适当的方法进行检查(例如 XRF、碱熔法 、其他酸 消解法等),以确认无目标元素损失。
5. 校准
5.1 校准溶液的制备
在逐级稀释每一种元素储备溶液后,将含有 0 μg~ 100 μg 每种元素的储备溶液转移到 100 mL 容量瓶[2.2 e)3)]中。若采用AFS法或内标法,则加入各种试剂,如适量的钴溶液[1 u)]和硫脲溶液[1 s)]、或掩蔽剂[1 u)]、或内标溶液[1 w)],使其与样品溶液中的试剂浓度相同。使用AAS法,可以是混合校准溶液。
5.2 校准曲线的建立
光谱仪用于定量分析。采用AAS 方法时,5.1 所得的样品溶液被雾化后引入氩等离子体或乙炔/空气火焰。当样品溶液含有 HF 时,应使用具有耐 HF 的进样系统。若采用 AFS法,测试溶液中的 Pb( II)被铁氰化钾氧化成 Pb(Ⅳ),然后与 KBH4 反应,生成挥发性氢化物 PbH4 或测试溶液中的 Cd 离子与 KBH4 反应生成挥发性气体,然后将 PbH4或气态 Cd 从液体中分离出来,用载气(Ar)引入石英炉中原子化。
a.AAS
——测得读数为目标元素的吸光度。在校准曲线法中,绘制目标元素吸光度与浓度之间的关系曲线作为校准曲线。
——在标准加入法中 ,将校准加入样品溶液中,通过将校准曲线外推至零吸光度来确定目标物未知浓度。
——表1 中给出了元素的典型测量波长供选择。如果存在共存物质的干扰,则应采用标准加入法。
b.AFS
——测定 Pb 时,应使用载流 1[1 r)l)]和氧化还原剂[1 p)]。测定 Cd 时,应使用载流2[1 r)2)]和还原剂 1[1 q)1)];测得读数为目标元素的荧光强度。在校准曲线法中,绘制目标元素荧光强度与浓度之间的关系曲线作为校准曲线。
——在标准加入法中,将校准标准系列加入样品溶液中 .通过将校准曲线外推到零荧光强度来确定目标物未知浓度。
校准曲线建立后,测定实验室试剂空白和样品溶液。如果样品浓度超出校准曲线的范围则应释溶液到校准曲线的范围内,确保酸度匹配,并进行再次测定。
测定的准确度通过使用标准物质、校准溶液等进行定期检查(例如每 10 个样品一次)。如果需要,应再次建立校准曲线。
在校准结果与预期值相差超过 20%的情况下 ,则应重新校准测定并重新测定该批次的所有样品。
如果将样品稀释到校准曲线范围,则应确保稀释的样品溶液中的酸度、内标和其他试剂浓度与储备溶液相当。
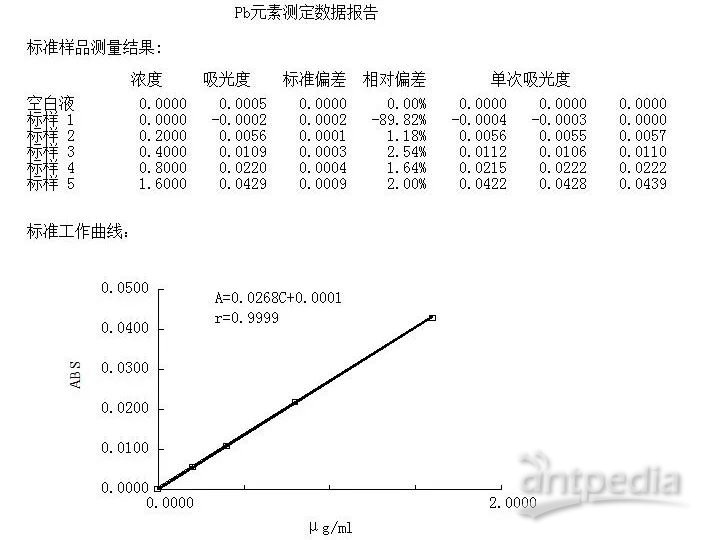
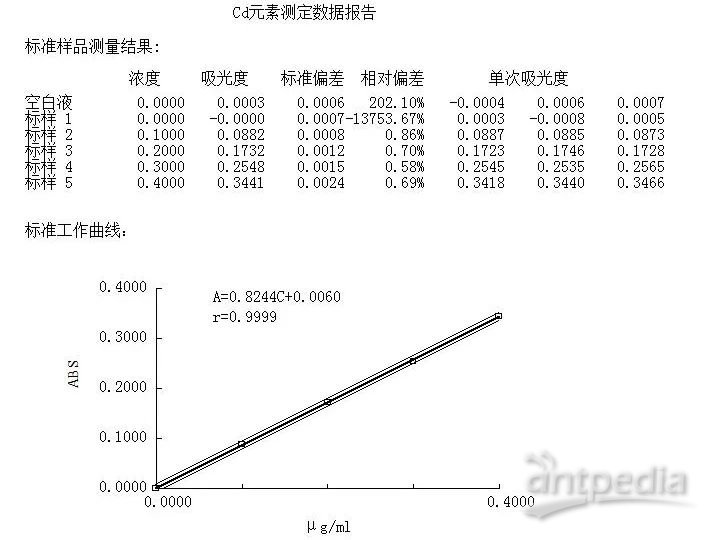
原子吸收法曲线:
6
6. 计算

式中:
c 样品中 Pb、Cd、Cr 的含量 ,单位为微克每克(μg/g);
A1——样品溶液中 Pb、Cd、Cr 的质量浓度 ,单位为毫克每升(mg/L);
A2——实验室试剂空白液中 Pb、Cd、Cr 的质量浓度 ,单位为毫克每升(mg/L);
V ——样品溶液的最终体积,取决于样品制备的稀释比例,单位为毫升(mL);
m ——所测样品的质量,单位为克(g)。
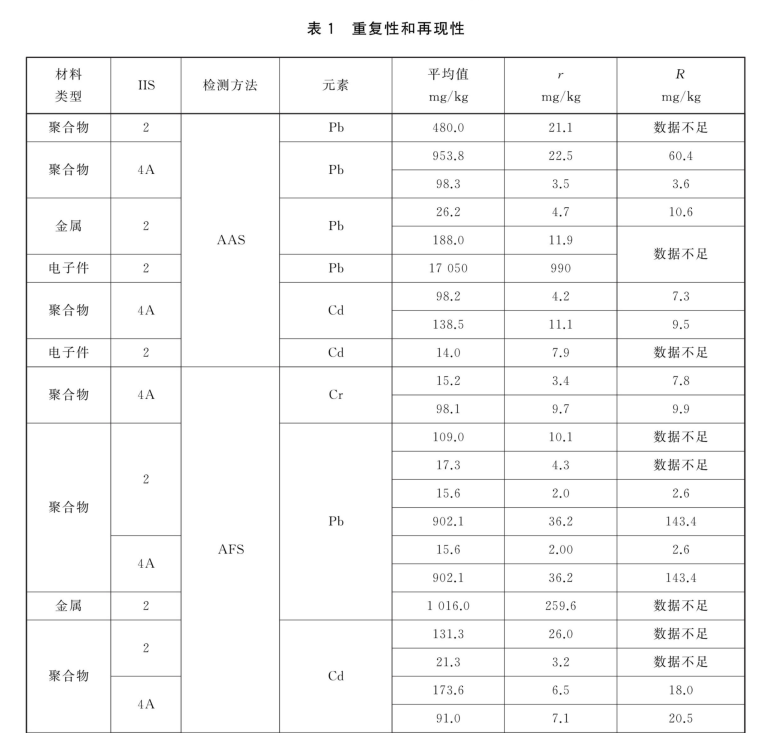
7. 精密度
在同一实验室内,由同一检测人员,使用相同的方法 ,相同的设备,在短时间间隔内.测试同一材料,当两个独立的单一检测结果的值在下面表 1 中所引用的平均值范围内时,超过 5%的情况下所获得的两次测试结果之间的绝对差异不超过国际实验室间研究 nos.2 (IIS2) 和 4A (IIS 4A)的统计分析得出的重复性限厂。
8. 质量控制
8.1 概述
适用时,每个测试方法标准的质量保证和控制要求应包括对质控样测试频率和验收准则的要求。本条款还应包括通用的质量控制方法如检出限(LOD)和定量限(LOQ)的确定。使用时,检出限和定量限应与 8.2 的描述一致。表 2 中列出了其他方法专有的质量控制要求 ,包括有关初始校验、方法空白、实验室控制样(LCS)等。

a) 每当建立校准曲线时,用一来自不同校准标准系列的标准进行初始校验。
b) 每批样品进行一次方法空白分析。不含铅 、镉或铬的空白基体可以作为方法空白样品。
c) 每批次进行的一个实验室控样(LCS)和实验室重复性试验 ,应通过在空白基体中添加铅、镉或铬的方法来分析。或者,重复检测含有铅 、镉或铬的有证标准物质。
d) 每测试 10 个样品和每批样品结束时,进行一次连续校验分析(CCV)。铅、镉或铬的回收率应在 90% 和 110%之间。如果连续校验分析(CCV)时铅、镉或铬的回收率超出此范围,则应在12 h 内对该 CCV 标准重新分析。如果对 CCV 标准进行再分析后,回收率仍然超出范围,则应停止分析,并对系统进行维护以使其恢复到最佳操作条件。最后一个正确的 CCV 校准结果前检测的样品都可以出具报告,但是 CCV 标准不符合之后的所有样本应用新的校准手段来进行重新检测。
按下列实验步骤确定铅、镉或铬的方法检出限和定量限。
a)准确称取适量已知不含铅、镉或铬的样品(例如 ,有证标准物质)或根据第 4 部分中的相关步骤可
能会干扰分析的其他化合物。将样品依次放入每个容器中,重复样品至少 5 个。
b) 在每个容器中加入 10 铅、镉或铬储备溶液[1 v)]。
c) 根据第 4 部分的测试相关步骤进行消解和光谱测定。
d) 按照第 6 部分的规定计算每种元素的含量μg/g),并按下列公式每个样品中加标元素的回收率百分比:

式中:
SR——以百分数表示的铅、镉或铬的加标回收率,%;
C——测得的含量 ,单位为微克每克(μg/g);
M——样品质量,单位为克(g);
SA——加标量(10 μg)。
每种样品的元素加标回收率应在 70% 125%之间。如果任一重复样品的加标回收率不在规定范围内,则应重复整个萃取和分析过程。
e) 方法检出限通过计算重复分析(至少 6 次)的标准偏差 s 来获得。然后,将标准偏差乘以学生 t值,重复检测 n 次时的自由度为 n 一1。表 3 给出了重复检测 6 到 10 次时的学生 t 值。
例如:重复检测 6 次时 ,自由度为 6—1=5,t值为 3.36。
注意:用于计算 MDL 的所有分析过程应是连续的。

f) 方法检出限乘以因子 5 为方法的定量限。
不同实验室的方法检出限和定量限可能不同。一般情况下,使用本方法测试都能达到 2 μg/g 的方法检出限(定量限为 10 μg/g)。
(部分资料来源于网络)


CAAM-2001系列原子吸收分光光度计

AFS-200型 顺序注射原子荧光光度计

TS-5双光束紫外可见分光光度计