







离子色谱(IC) ICP-MS 分析天然水中的铬
摘要
痕量元素铬的分析在很多不同基体物质分析应用中非常热门。铬在环境中以两种不同的氧化态存在,即三价Cr(III) 阳离子和六价Cr(VI) 阴离子。在哺乳动物体中,Cr(III) 在葡萄糖调节过程中是一个基本元素。然而,已证明较低含量水平的六价铬形式具有诱导有机体突变和致癌效应。由于铬元素的这种二元性,总铬浓度并不能提供充分的信息以确定其潜在的毒性。为了评价样品的潜在毒性,不能仅测量其总浓度,必须测量Cr(VI) 的含量。本工作采用离子色谱和八极杆反应池ICP-MS联用技术,建立了一个简单可靠的测定Cr(III) 和Cr(VI) 的方法,为样品中有毒铬的含量提供了准确信息。该方法由于样品制备和色谱条件的最优化,可以分析高基体样品,比如硬质饮用水。另外,由于反应池中消除了一些潜在的背景干扰离子,ICP-MS 方法提供了卓越的信噪比,可以准确测量对于毒理学有用的Cr(VI) 含量,即低于0.1 μg/L 的浓度。
引言
需要检测铬毒性的样品种类范围很宽,包括饮用水、食品以及临床样品(临床样品主要用于职业暴露评价)。不过,有毒的只是铬的六价形式Cr(VI) ,而三价形式的铬则是人体营养的基本元素。因此,要想确定铬的毒性,必须测量其Cr(VI) ,而不是简单的总
铬浓度。有两个途径可以解决这个问题:首先,如果测量的总铬浓度低于有毒的Cr(VI) 水平,那么即使所有的铬都是以Cr(VI) 存在,也有理由得出该样品中铬的浓度不具有毒性。不过,这种方法对于含有高浓度Cr(III) 的样品来讲,会导致大量的假阳性现象,因此比较准确的方法是分离后测定Cr(VI) 本身。理想的方法是对两种铬的形态分离和测定,一次分析可以得到总铬和有毒的Cr(VI) 的含量。元素的不同形态的分离和检测通常是一个直面的挑战。而铬的形态分析却不寻常。这是因为天然样品,比如水中铬的一般形态是Cr(VI) 的铬酸盐(CrO42–) 以及Cr(III) 的铬离子(Cr3+)。铬酸盐是一种阴离子,而三价铬离子是阳离子。所以在相同条件下,采用一种离子交换方法是不能对这两种离子都起作用的。还有一个问题是在诸如水这样的样品中,Cr(III) 是最稳定的氧化态,而Cr(VI) 离子则是强氧化剂,在酸介质或有机物存在时,很容易被还原。因此,在样品采集、储存和制备过程中都必须非常小心,以保证原始样品中铬的形态分配在分析之前保持不变。因此,在样品采集、储存和制备过程中都必须非常小心,以保证原始样品中铬的形态分配在分析之前保持不变。
实验
本文介绍的方法采用一种最优化的样品稳定方法,将样品与EDTA 放在40 °C 恒温箱中,EDTA 与Cr(III)形成一种络合物,然后用一个简单的色谱法分离Cr(III)EDTA 络合物和Cr(VI)。形成Cr(III)EDTA 络合物的反应取决于保温时间和温度。在60 °C 1 个小时之内或40 °C 3 小时条件下,反应完全。在室温条件下,即使放置7 小时,反应也不会完全。注意该方法对于天然水样品,需要加入比较高浓度的EDTA。因为其它一些离子,比如Ca 和Mg,通常在硬质饮用水中的浓度为10’s 或100’s mg/L,也会和Cr(III) 竞争形成EDTA 的络合物,如EDTA 量不够,导致Cr(III) 的回收率随基体偏低。采用离子色谱(IC) 分离Cr 的形态,ICP-MS 测定的联用技术是一种理想的分析方法。用一种简单的、低成本的离子色谱装置就可以分离铬的不同形态。ICPMS可以实现相当低浓度的铬形态的测量,提供准确的暴露水平评价浓度,甚至可以测量天然的或背景水平浓度的铬。ICP-MS 具有卓越的灵敏度,因此是许多元素很好的检测器。碰撞/反应池的引入,消除了基体有关的ArC 和ClOH 的干扰,所以ICP-MS 可以更为准确和灵敏的测定主同位素52Cr。铬形态分离所用的样品制备方法,色谱柱类型以及色谱条件列于表1。注意,除了样品用EDTA 钠盐稳定外,流动项中也要加入EDTA,在分离期间稳定Cr(III) 的络合物。另外,必须将溶液调到pH 7,以利于铬形态的稳定和最优化的色谱分离。

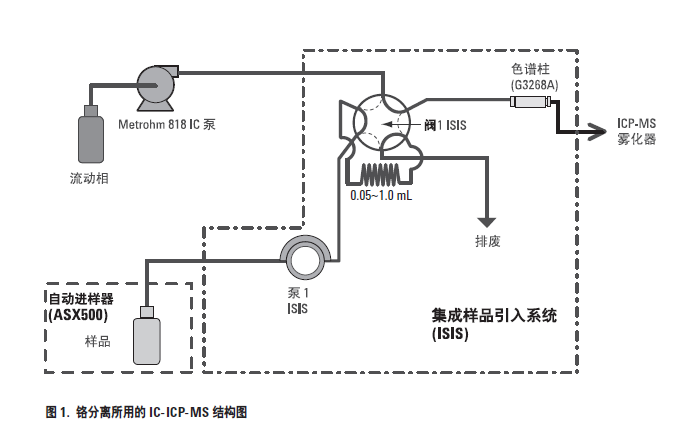
图1 是本工作所用的离子色谱的结构。注意,采用非金属离子色谱泵(Metrohm 818 IC 泵)传输流动相,用安捷伦7500ce ICP-MS 的可选集成样品引入系统(ISIS) 填载和切换样品环。这种结构维持高精密度和较高的IC 泵压力,比完整的IC 或HPLC 系统更为简单且成本低。因为除了ICP-MS 项系统,只需要配上IC 泵模块即可。
结果和讨论
在上述条件下,用安捷伦7500ce 的H2 池模式去除ArC 和ClOH 对52Cr 的干扰,铬形态分析的检出限(DLs) <20 ng/L,见表2。许多有关六价铬的国际规范中指定的最大允许浓度是1 μg/L,要求的检出限是其十分之一(100 ng/L)。本工作即使采用少至100 μL的进样体积,也还是很容易满足这些要求的。不过,将进样体积增加到500 μL,检出限降低到13.2 ng/LCr(III),15.8 ng/L Cr(VI)。
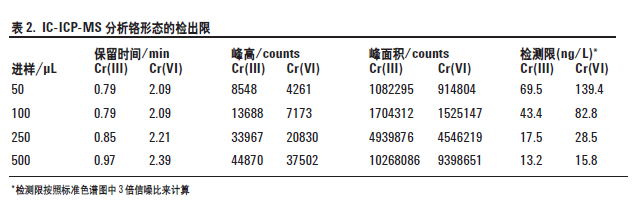
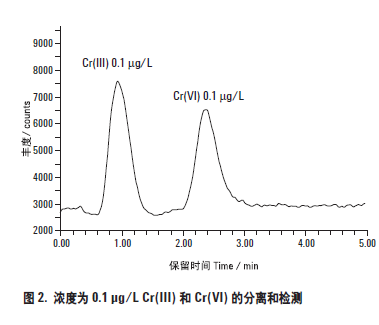
如图2 所示,对于这些条件下简单的标准溶液,两种铬形态的信噪比都很好。该色谱图给出了浓度为0.1μg/L (ppb),进样体积为500 μL 的两种铬形态的分离情况。从图中可以看出,在总时间大约3 分钟时,两种铬形态的峰很容易在背景和分离基线以上被检测。
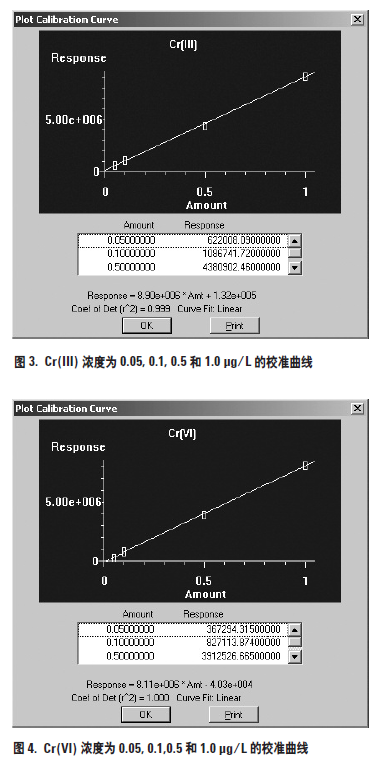
用低浓度的人工合成标准溶液建立了不同铬形态的校准曲线。定量分析基于峰面积测量。Cr(III) 和Cr(VI) 的校准曲线分别示于图3 和图4。两个校准曲线都具有极好的灵敏度和线性。
对于适合于常规分析的方法,除了灵敏度外,形态的稳定性、色谱分离以及校准的线性,要求能提供可接受的长期稳定性。在色谱分析中,稳定性受控于两个因素,即保留时间RT 和峰面积的稳定性。表3 的数据证明了这些参数的稳定性,说明该方法的稳定性可以被常规分析所接受。
常规分析
对于天然水样品中铬形态的常规分析来讲,更为重要的是在含有高浓度的其它离子的实际样品中能够保持这种极好的灵敏度、稳定性和色谱分离。为了检验方法对真实样品的的适应性,用该方法测定了加标和未加标的矿泉水样品中铬的两种形态。第一个用于评价的样品是一个有名的法国矿泉水,本研究标示为矿泉水A。图5 是对这个矿泉水A 加标和未加标的两种铬形态的色谱图。水的主要因素成分也示于图的插入表格中。一般饮用水大约含100 mg/LCa ; K, Mg 和Na 等其它元素在5 mg/L 和20 mg/L之间。
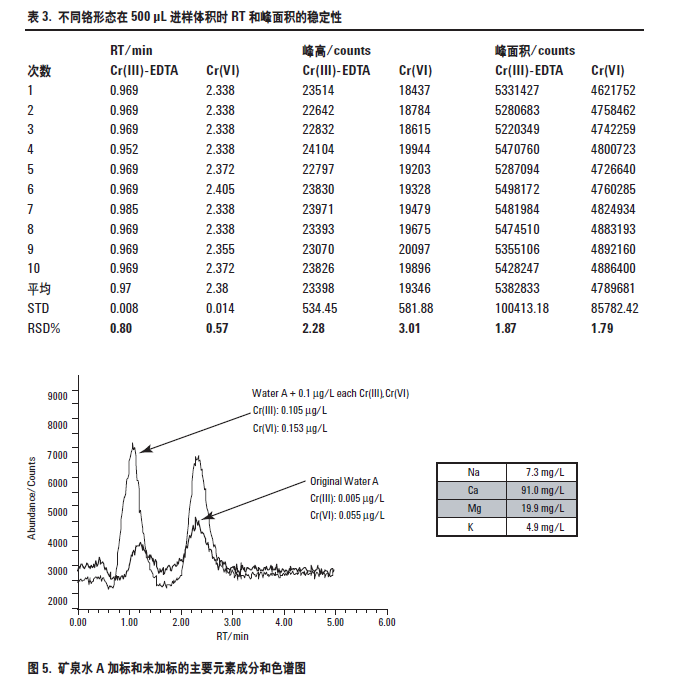
矿泉水A 的加标回收率示于表4。从表中结果可以看出,铬形态定量分析的浓度很低(对于Cr(III) 和Cr(VI) 分别为0.005 μg/L 和0.055 μg/L),这个样品的低浓度加标回收率很好,两种形态分析的误差小于5%。

第二个矿泉水样品也是一个法国矿泉水,标示为矿泉水B。该矿泉水与一般矿泉水相比,含钙和硫酸盐较高(超过450 mg/L Ca;大于1000 mg/L 的硫酸盐)。和矿泉水A 一样,也进行了加标和未加标实验。分析得到的色谱图示于图6,加标回收数据列于表5。
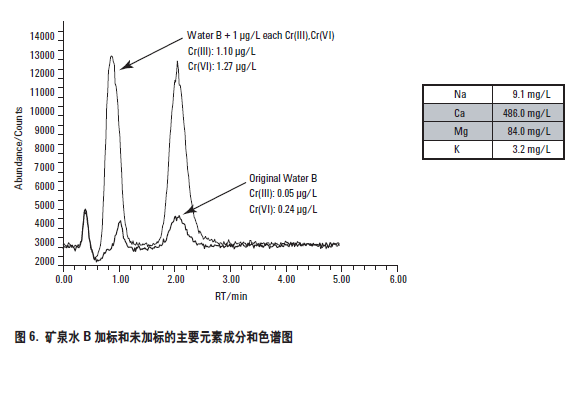
如同矿泉水样品A,矿泉水B 的主要元素成分示于色谱图的插入表中,可以看出其中的矿物质含量很高。尽管如此高的主要元素存在,但由于采用了最优化的样品制备和色谱方法,仍然获得了很好的色谱分离和极好的加标回收率结果。在这个样品中,由于铬的基础浓度(未加标)较高,所以采用了较高的加标量。高基体样品中低浓度铬形态的回收能力证明了样品稳定过程所采用的最优化方法的有效性,即加入了足够浓度的EDTA 以保证即使存在高含量的其它竞争离子,Cr(III) 也能被完全络合。此外,两种铬形态在低浓度的准确加标回收率表明,通过对样品稳定化期间以及流动相选择合适的pH,避免了可能存在的形态之间的转化问题(Cr(VI) 还原为Cr(III))。见表5。
结论
建立了一个天然高基体水样中Cr(III) 和Cr(IV) 的最佳稳定化和测定的新方法。检出限在100-μL 进样体积时为0.05 μg/L;500-μL 进样体积为0.015 μg/L。每个样品可靠稳定分离Cr(III) 和Cr(VI) 需要大约3分钟的时间。对于含有高浓度竞争离子的样品,比如>500 mg/L 的矿物元素的矿泉水,也能够获得准确的形态分离和定量分析结果。采用Agilent 7500ce 的氢气反应池模式,可以实现国际规则所要求的低浓度铬(0.1 μg/L)的准确无干扰分析。











