捕集离子淌度质谱带来磷酸化蛋白质组研究新的深度
Yasushi Ishihama 教授
京都大学分子与细胞生物分析实验室
2021年1月发表于www.ddw-online.com
目前,在蛋白质生物化学和蛋白质组学相关研究中,质谱(MS)已广泛应用于鉴定和表征蛋白质。且随着技术的不断发展,质谱已经实现了更高的覆盖深度、更快的分析速度和更高的灵敏度。例如,串联质谱(MS/MS)是一种灵敏、准确、有效的检测手段,通过产生并检测序列特异性碎片离子1对肽段进行解析。同时,相较于传统的PTMs分析方法,如埃德曼降解法、氨基酸分析法、同位素标记法、免疫化学法等,存在如氨基酸序列覆盖不完整,对不同PTMs的串扰、选择性及其对细胞调控网络的影响等诸多问题。串联质谱也可作为解析蛋白质翻译后修饰(PTMs)的有效工具,用于阐明控制细胞活动的复杂过程,如细胞分裂、生长和分化。
在可逆PTM中,蛋白质磷酸化为最主要的形式之一,用以调节细胞重要的代谢、激素、发育和应激反应,因此,在细胞的蛋白质合成、细胞分裂、信号转导、细胞生长、发育和衰老2过程中,起着至关重要的作用。磷酸化水平异常会导致如癌症和糖尿病等一系列疾病,因此,磷酸化蛋白质组学成为了探索健康和疾病密码的重要工具。在细胞信号转导网络中,可逆磷酸化是将信号转导到细胞核以控制基因表达的其中一项关键因子。此前研究学者估计人体内约有30%的蛋白质被磷酸化,但京都大学分子和细胞生物分析实验室的研究人员开发了一种高选择性的磷酸肽富集方法,并将该方法应用于检测细胞磷酸化,研究结果发现:人体内至少有70%的蛋白质被磷酸化,超出了原先的预估3,4。
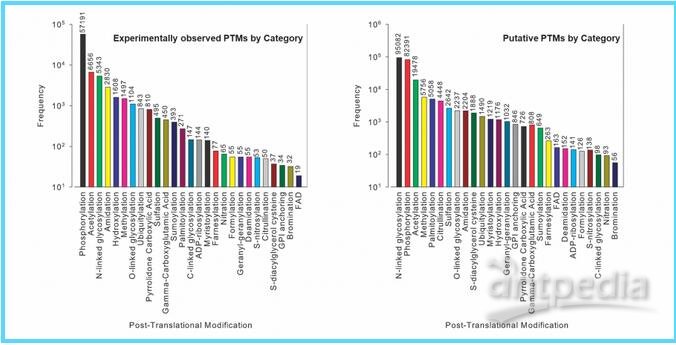
图1:Swiss-prot中各种PTMs统计概览(参考文献:Khoury, G., Baliban, R. & Floudas, C. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep 1, 90 (2011))
捕集离子淌度质谱已经成为磷酸化蛋白质组学研究最强大的工具
随着技术的进步,人们逐渐在磷酸化蛋白质组学方法中使用质谱技术来研究蛋白质磷酸化。通过对蛋白质和PTM的深入表征和定量分析,人们发现,理解蛋白的信号传导途径和异常疾病状态至关重要。高分辨质谱仪的发展和专为蛋白质磷酸化整体分析量身定制的磷酸化肽富集技术,为分子和细胞生物学家研究蛋白信号转导途径提供了强大的工具。尽管人们已经取得了一些进展,但与未修饰的肽段检测相比,识别PTM仍是一项挑战。由于PTM蛋白通常以低丰度形式出现,并且不同蛋白质磷酸化的差异极大(跨多个数量级),由此推动了人们对更高灵敏度、更高峰容量检测仪器的需求。
离子迁移谱(IMS)是一种气相后电离分离方法,当与质谱结合使用时,可基于结构(离子构象)和质荷比(m/ z)对分析物进行快速、高分辨率的分离5。IMS-MS是一项完善的技术,在蛋白质分析方面已显示出巨大的潜力,他可以提升多肽的鉴定质量,提供与LC-MS质量信息互补的结构信息。IMS-MS根据离子的形状(IMS)和质量(MS)的差异来分离离子,从而传递离子的三维(3D)结构信息。这种通过构象差异分离离子的能力使得人们分离同分异构体成为可能,例如磷酸肽位置异构体(即肽段中磷酸化数目相同,但位点不同),而传统质谱技术很难将它们区分6。
2017年,通过特定质量和迁移率的离子累积和聚焦的离子淌度技术,即捕集离子淌度(TIMS)技术被引入蛋白质组学,这使蛋白质分析的灵敏度和速度有了更进一步的革命性增长。TIMS能够以毫秒为单位,在气相中以高效、短循环周期和高分辨率特性对离子迁进行淌度分析,并且使用测量其碰撞截面积(CCS)7,并将CCS值用于蛋白质组学工作流程中,从而产生了由质量、强度、保留时间和CCS的4D-蛋白质组学方案。
TIMS通常与飞行时间质谱(TOF)结合使用,以达到更高的分析速度。结合平行累积连续碎裂(PASEF)技术,TIMS-QTOF MS系统可以提供更高速、更高灵敏度的检测方法。新颖的设计使离子可以在系统前部淌度分析器进行离子的累积和聚焦,而后部淌度分析器则根据其离子迁移率,透过调控电场将离子依次释放。TIMS与PASEF的强大结合可在少量样品情况下,实现更大的磷酸化蛋白质组覆盖率,并且通过使四级杆与TIMS,在特定肽段的洗脱时间内,提供>100Hz的测序速度而不损失灵敏度或分辨率。
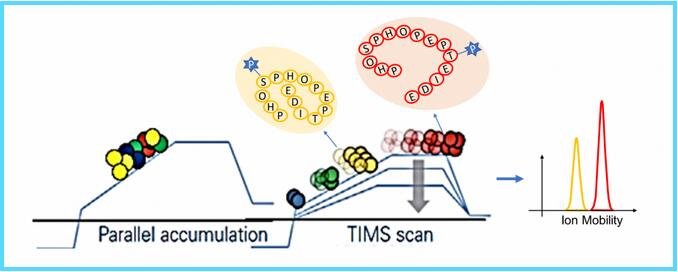
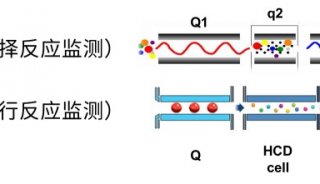
图2:捕集离子淌度用于磷酸化修饰位点区分
例如,京都大学的研究小组发现PASEF可以有效地提高串联质谱的采集速率,从而达到磷酸化蛋白质组学研究新深度,提高鉴定结果的可靠性。PASEF采集模式的高速度和高灵敏度也有助于TIMS-QTOF MS实现高通量分析。这种优势对宏蛋白质组学的相关研究极为重要,如分析大量样品以在蛋白质组水平上研究人类和细菌之间的相互作用。下一步便是在现有的代谢蛋白质组学研究中利用PASEF的高速采集模式,找寻与疾病相关的潜在生物标志物8。
捕集离子淌度质谱用于激酶信号转导研究
激酶信号是通过可逆酶促反应在底物上加上磷酸基团产生的,是细胞活动的重要组成部分,详细研究激酶信号过程对于理解众多疾病和开发新疗法至关重要9。激酶介导的磷酸化信号通路是已知导致或驱动如癌症等疾病发展的重要因素。京都大学的研究小组已经开发了磷酸化蛋白质组学方法,对激酶靶向药物的体内磷酸化蛋白质组分析,其目的是促进癌症治疗药物的发现和开发,进而探索、分析磷酸化分子的功能。因此,TIMS-QTOF-MS在药物发现中,特别是在与癌症相关的磷酸化方面具有重要意义。目前,人们已经开发出许多抑制特定激酶的药物。迄今为止,已有超过19000种针对约260种蛋白激酶的激酶抑制剂被报道10,约30种小分子激酶抑制剂已被美国食品和药物管理局(FDA)批准用于临床11。
京都大学的研究小组通过使用timsTOF Pro(布鲁克)实现1分钟梯度洗脱,并将其用于研究激酶抑制剂的作用机制(图3)。这项工作希望通过实验和计算,将激酶与其底物相结合,以揭示整个信号网络。
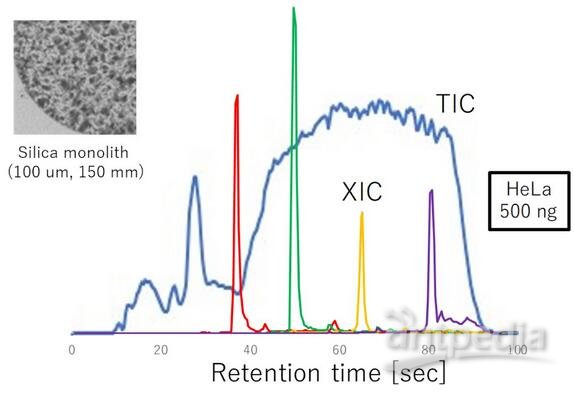
图3:带有timsTOF Pro的毛细管LC 1分钟梯度,100 s/run,864 run/d TIC =总离子色谱图;XIC =提取离子色谱图。
在过去的20年里,基于质谱的磷酸化蛋白研究已被应用于许多大规模分析细胞信号转导的研究中。现在,基于LC-MS/MS12,13上进行的激酶组研究,研究人员现在正在达到只有使用TIMS-QTOF MS才能达到的通量水平。
持续的技术创新推进蛋白质组学研究
显然,基于质谱的蛋白质组学研究的发展(如使用TIMS技术的4D-蛋白质组学),已经让研究人员在高通量、高灵敏度条件下深入了解分子和细胞功能。特别是使用PASEF采集法提供的超快分析速度和超高灵敏度,在低样本量条件下就能达到鸟枪蛋白质组学和磷酸蛋白质组学研究新深度。这种4D-蛋白质组学方法将使科学家能够在常规情况下探索以前无法获得的蛋白质信息。
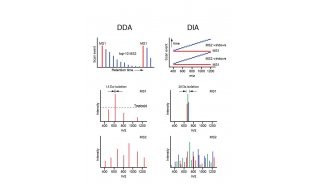
各种计算蛋白质组学的方法推动了蛋白质组学的未来发展,数据非依赖型采集(DIA)方法是基于质谱技术的磷酸化蛋白质组学的重要进展。DIA是近来较新开发的一种质谱采集技术,相比于数据依赖型采集(DDA),DIA以连续、无偏的方式对特定质量范围内的所有前体离子进行MS2扫描。二代测序(NGS)技术与蛋白质组学的结合,在蛋白质基因组学等多组学领域,尤其是肿瘤临床研究中得到了广泛应用14。
通过将DIA与PASEF相结合,研究人员可以通过TIMS和m/Z双重隔离模式来弥补传统的DIA缺陷(图4)。在dia-PASEF方法中可以大大增加离子利用百分比(对于低复杂度样品能达到100%,对于高复杂度的样品,仍然比使用类似隔离模式大小和m/z范围的传统DIA方法高5倍)。这可以缩短dia-PASEF采集的循环时间,使其与短梯度分离兼容,又能保持高选择性。受益于TIMS空间聚焦效应,dia-PASEF不仅提高了检测灵敏度,在全面4D-蛋白质组学支持下,可以将DIA选择性提高到一个新水平。
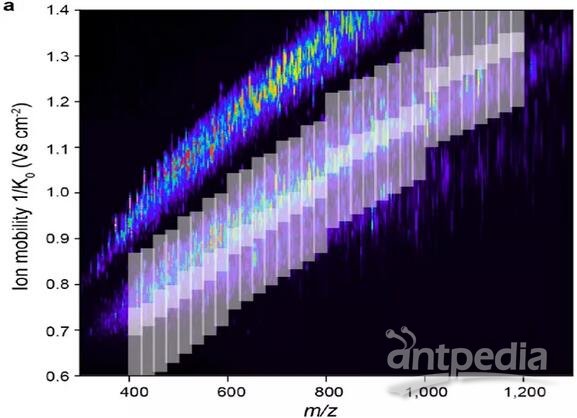
图4:dia-PASEF窗口分配示例。使用25Da宽的64个窗口,1.7秒循环时间dia-PASEF方法。此方法使用6.25%的离子,等效的3D DIA方案仅使用1.25%。
在过去的二十年中,质谱技术和新方法(例如TIMS,PASEF和dia-PASEF)已取得了重大进展,并将磷酸化蛋白质组学转变为蛋白质研究者、生物学家和临床研究人员寻求对蛋白质和细胞信号的更深入了解,并将其转化为疾病转化模型的强大工具。
参考文献
1. Larsen MR, Trelle MB, Thingholm TE and Jensen ON(2018) Analysis of posttranslational modifications of proteins by tandem mass spectrometry, BioTechniques, 40(6): 790-798.
2. Ardito F, Giuliani M, Perrone D, Troiano G, LoMuzio L (2017) The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int J Mol Med. 40(2):271-280.
3. Olsen JV, Vermeulen M, Santamaria A, Kumar C,Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M(2010) Quantitative phosphoproteomics reveals widespread full phosphorylationsite occupancy during mitosis, Sci Signal, 3(104):ra3.
4. Sharma K, D’Souza RCJ, Tyanova S, Schaab C, Wiśniewski JR, Cox J and Mann M (2014) Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr andSer/Thr-Based Signaling, Cell Reports, 8(5):1583-1594.
5. McLean JA, Ruotolo BT, Gillig KJ, and Russell DH(2005) Ion mobility-mass spectrometry: a new paradigm for proteomics, International Journal of Proteomics. 240(3): 301-315.
6. Glover MS, Dilger JM, Acton MD, Arnold RJ, Radivojac P, and Clemmer DE (2016) Examining the Influence of Phosphorylationon Peptide Ion Structure by Ion Mobility Spectrometry-Mass Spectrometry, J AmSoc Mass Spectrom, 27(5): 786-794.
7. Fernandez-Lima F (2016) Trapped Ion Mobility Spectrometry: past, present and future trends, J. Ion Mobil. Spec, 19:65–67.
8. Lin, Miao-Hsia; Sugiyama, Naoyuki; Ishihama,Yasushi. Systematic profiling of the bacterial phosphoproteome revealsbacterium-specific features of phosphorylation. Sci Signal,8(394):rs10.
9. Savage SR and Zhang B (2020) Using phosphoproteomics data to understand cellular signaling: a comprehensive guideto bioinformatics resources, Clinical Proteomics, 17:27.
10. Hu Y, Furtmann N and Bajorath J (2015) Current compound coverage of the kinome, JMed Chem, 58(1):30-40.
11. WuP, Nielsen TE and Clausen MH (2016) Small-molecule kinase inhibitors: ananalysis of FDA-approved drugs, Drug Discovery Today, 21(1):5-10.
12. SugiyamaN and Ishihama Y (2016) Large-scale profiling of protein-kinases for cellular signaling studies by mass spectrometry and other techniques, Journal of Pharmaceutical and Biomedical Analysis, 130: 264-272.
13. Sugiyama N, Imamura H and Ishihama Y (2019) Large-scale Discovery of Substrates of the Human Kinome, Scientific Reports, 9:10503.
14. Ang MY, Low TY, Lee PY, Nazarie WFWM, Guryev V and Jamal R (2019) Proteogenomics: From next-generation sequencing (NGS) and mass spectrometry-based proteomics toprecision medicine, Clinica Chimica Acta, 498:38-46.



 400-6699-117 转 2005
400-6699-117 转 2005