蛋白质组质谱新方法一览
2003年人类基因组精细图绘制完成,是人类科学史上一个里程碑式的事件。后基因组时代的研究重点自然落在了蛋白质头上。为啥?因为中心法则告诉我们,基因的产物——蛋白质,是生命活动的最终执行者。与基因组类比,研究生物体内全套蛋白质的科学,就是蛋白质组学。基因组计划完成的同年,人类蛋白质组计划启动,令人激动的是,2014年人类蛋白质组的草图也完成了。而蛋白质组学能够飞速发展的最大功臣非质谱莫属。质谱的应用范围非常广泛,但这里只讨论蛋白质组学中的质谱。
简单地说,质谱法(mass spectrometry)就是对肽段离子的重量(质荷比,m/z)进行测量的分析方法。样品经质谱仪(mass spectrometer)检测得到质谱图(mass spectrum),通过对质谱图的分析就可以对样品中的蛋白进行鉴定、定量。亲,图1的这种典型的蛋白质组学流程都很熟悉吧。蛋白首先都要被特异性的酶(通常为Trypsin)切割为肽段,再进行后续分析,这在蛋白质组学中被称为“自下而上”的研究策略(Bottom-up proteomics)。我们平时见到的质谱分析基本都是这种类型。提到蛋白质组,即会联想到一系列高大上的名词,iTRAQ、SWATH、SILAC、Shotgun、Label-free等等。很多概念容易弄混淆,下面我们就来理理清楚。
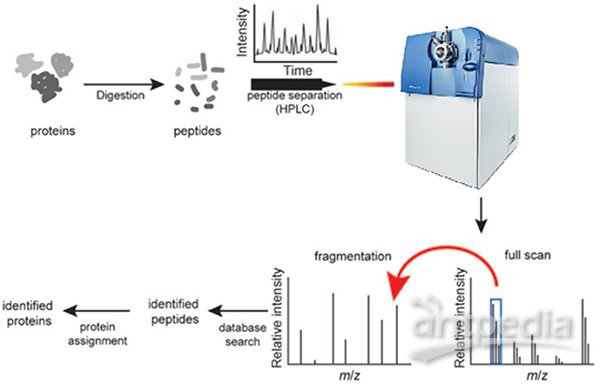
图1. 典型的蛋白质组学流程
大体上,质谱研究蛋白主要是鉴定和定量。通过二级质谱图(MS2或者MS/MS)进行数据库搜索匹配鉴定蛋白。通过各种标记或非标记的手段对不同样品中的蛋白进行比较就是定量。蛋白定量比较是质谱最重要的用途,图2是对定量方法的一个简单总结。
非标定量(Label-free)不需要标记,不同样品分别处理、分别进质谱检测;优点是处理简单、无需标记、价格便宜、可以比较很多组样品,缺点是对操作步骤、LC、质谱稳定性要求严格。
蛋白质非标记定量技术(label-free)是通过液质联用技术对蛋白质酶解肽段进行质谱分析,无需使用昂贵的稳定同位素标签做内部标准,只需分析大规模鉴定蛋白质时所产生的质谱数据,比较不同样品中相应肽段的信号强度,从而对肽段对应的蛋白质进行相对定量。特点:(1)无需昂贵的同位素标签做内部标准,实验耗费低;(2)对样本的操作少,从而使其最接近原始状态;(3)不受样品条件的限制,克服标记定量技术在对多个样本进行分析方面的缺陷;(4)对实验操作稳定性、重复性要求高,要求至少做三次生物重复。
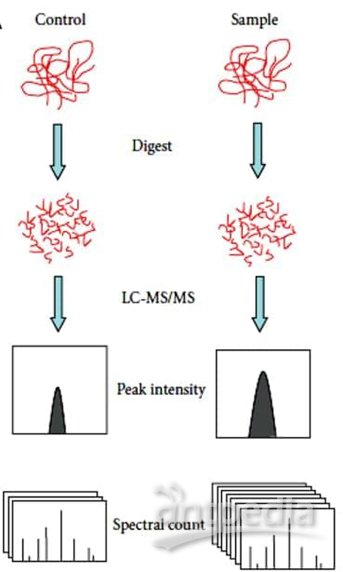
Lable Free定量(LFQ)
SILAC是在细胞培养基中加入稳定同位素标记的氨基酸,在代谢水平标记蛋白,一级质谱图进行定量,可以做到三组样品混合后进行比较,定量准确,但是不能标记组织样本,养细胞成本也较贵。
SILAC即细胞培养条件下稳定同位素标记技术(Stable Isotope Labeling By Amino Acids In Cell Culture,SILAC),其实验原理是:
•在细胞培养基中加入轻、中或重型稳定同位素标记的必需氨基酸(赖氨酸和精氨酸),通过细胞的正常代谢,使新合成的蛋白带上稳定同位素标签。
•等量混合各类型蛋白质,酶解后进行质谱分析。
•通过比较一级质谱图中同位素峰型的面积大小进行相对定量,
•同时二级谱图对肽段进行序列测定从而进行蛋白鉴定。
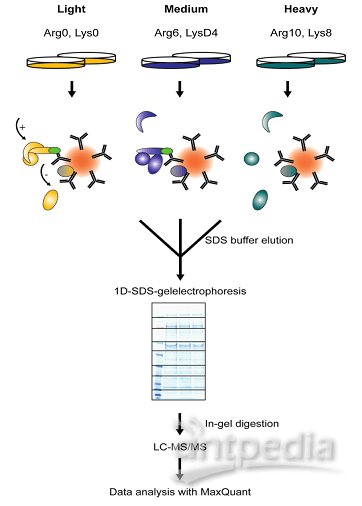
SILAC
双甲基化标记是通过化学反应的办法在肽段水平进行标记,一级质谱定量,也可以三组对比,标记试剂都比较便宜,而且可以标记任何来源的样品。其特征在于:利用作用位点为赖氨酸羧基的蛋白酶将蛋白质酶切成肽段,根据肽段的N末端氨基和C末端赖氨酸侧链氨基在pH酸性条件下反应速度不同的性质,依次在酸性、碱性条件下对肽段N末端和C末端进行二甲基化标记;通过二甲基化标记试剂甲醛CH2O、氰基硼氢化钠NaBH3CN及其相应同位素13CH2O、CD2O、13CD2O、NaBD3CN的组合实现两重至六重中一种或二种以上的标记,使肽段每端的标记有所不同,而肽段的总质荷比相同,从而使肽段在液质联用的质谱仪中得到的一级谱具有相同的质荷比,而二级谱每种碎片离子都存在质荷比差异,利用二级谱碎片离子强度进行蛋白质的多重定量分析。
按照权利要求1所述的定量方法,其特征在于:所述依次在酸性、碱性条件下对肽段N末端和C末端进行二甲基化标记,并通过二甲基化标记试剂甲醛、氰基硼氢化钠及其相应同位素的组合实现六重标记,六重标记试剂为:
肽段N末端:13CH2O+NaBH3CN——肽段C末端:CD2O+NaBD3CN;
肽段N末端:CD2O+NaBH3CN——肽段C末端:13CH2O+NaBD3CN;
肽段N末端:13CD2O+NaBH3CN——肽段C末端:CH2O+NaBD3CN;
肽段N末端:CH2O+NaBD3CN——肽段C末端:13CD2O+NaBH3CN;
肽段N末端:13CH2O+NaBD3CN——肽段C末端:CD2O+NaBH3CN;
肽段N末端:CD2O+NaBD3CN——肽段C末端:13CH2O+NaBH3CN。
双甲基化标记定量
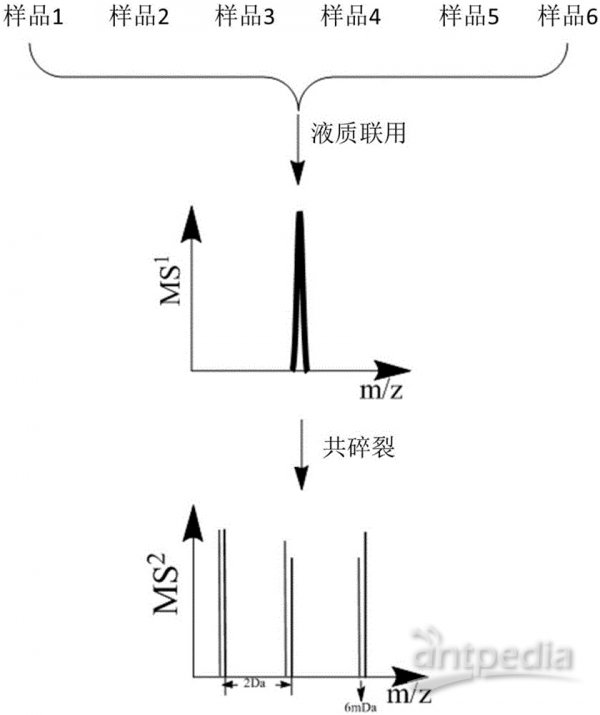
iTRAQ和TMT是商品化的试剂盒,肽段水平标记,二级质谱定量;分别可以做到最多8组和10组样品间蛋白质组的比较。
以iTRAQ为例(TMT和iTRAQ类似),iTRAQ是一种同位素标记试剂,可与氨基连接的胺标记同重元素。两种:4-plex和8-plex,可同时标记4组或8组样品。
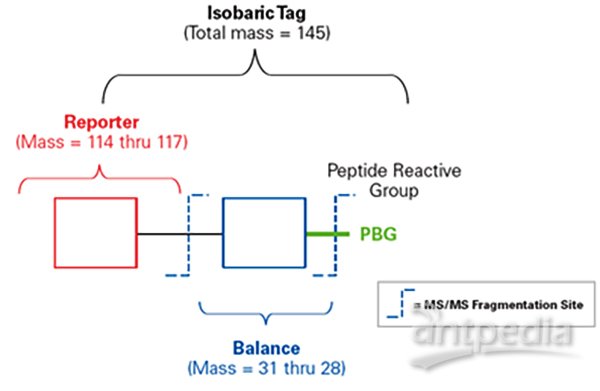
•iTRAQ包括三部分:报告基团(reporter group ),平衡基团(balance group),肽反应基团(peptide reactive group)。
•报告基团(reporter group):质量分别为114Da、115Da、116Da、117Da。
•平衡基团(balance group):质量分别为31Da、30Da、29Da、28Da,使得四种iTRAQ试剂报告基团和平衡分子的总分子量均为145Da,无论使用哪种iTRAQ试剂,不同同位素标记同一肽段后在一级质谱中,分子量完全相同,呈现的都是同一峰值。
•肽反应基团(peptide reactive group):将reporter group与肽N端及赖氨酸侧链连接,从而将报告基团和平衡基团标记到肽段上,几乎可以标记样本中所有蛋白质。
•8-plex的报告基团共有八种,质量分数分别为114-121Da,因此iTRAQ最多可同时标记8组样品。
分别标记各酶解后的样品,混合,一级质谱全一样,二级质谱各报告离子分开,对各二级质谱定量,即可知在各样品中的相对含量,即相对定量。
4-plex为例说明iTRAQ试剂标记原理
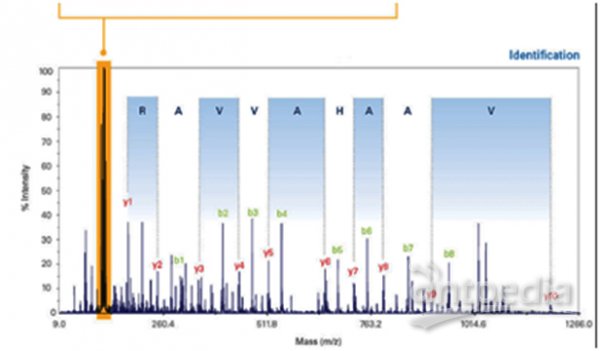
iTRAQ标记后,一级质谱全一样
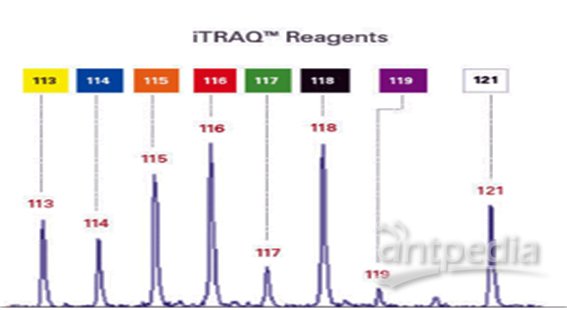
iTRAQ:利用二级质谱报告离子定量
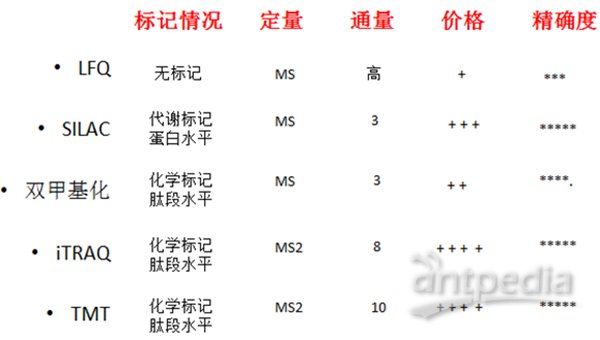
图2. 质谱定量方法
以上这几个是一家的,还有几个名词是属于另外一家,比如Shotgun (DDA)、SWATH/DIA、SRM (MRM)、MRMHR/PRM。质谱进行数据采集的方式大致分为三种:鸟枪法(Shotgun)、选择反应监控(SRM)和全景式的SWATH/DIA。下面对照图3再来简单介绍一下。
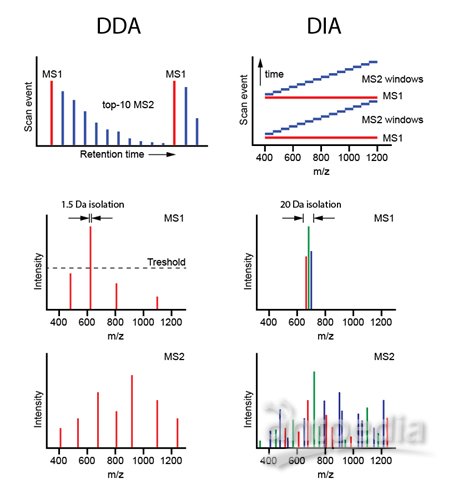
DDA(shotgun)和SWATH/DIA的区别
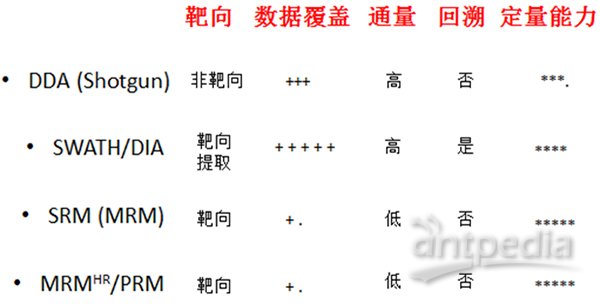
图3. 质谱扫描方式
DDA、IDA、Shotgun和鸟枪法说的是相同的东西,意思是质谱在每个循环的中从一级里挑选丰度高的TopN个肽段去打碎做二级扫描,得到的结果通过与已知数据库中的理论蛋白进行匹配。DDA简单有效,分析流程比较成熟,也是目前质谱分析的主流方式。DDA也有其固有的缺陷,即具有一定的随机性,偏向于检测丰度较高的肽段,而抑制了低丰度肽段的检测。
靶向策略被称为质谱领域的Western blot。质谱只去采集目标肽段大小的离子信息,因而提高了灵敏度和特异性。这种方法用来研究感兴趣的特定蛋白,定量准确,但是通量很有限。
SWATH/DIA这种全景式的数据采集方式在最近几年突然火了起来,被认为在不远的未来可能会取代DDA的主流位置。该方法采取的策略是将扫描范围内的所有肽段按照质荷比分为若干个窗口,再对每个窗口里所有的肽段一起打碎,采二级,数据分析时通过抽提蛋白的子离子信息进行定量。SWATH/DIA解决了DDA中随机性选择肽段的缺陷,所以重复性更好,定量的准确性基本达到了SRM的水平,而且可以实现大规模定量。
借用听来的一个比喻来说明:DDA就像机关枪扫射,数量多、体积大的目标命中的概率要大一些。靶向扫描(SRM或PRM)就像精准狙击,排除干扰,目标明确,每一枪直指目标,但是难以大规模消灭敌人。SWATH/DIA就是地毯式轰炸,只要暴露在我方攻击范围内的敌人,不管三七二十一,全部炸完。
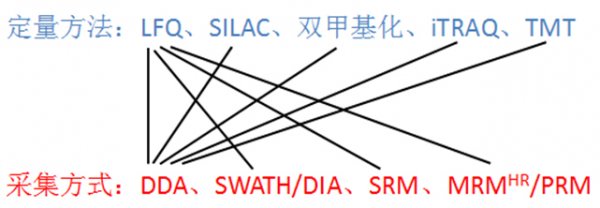
图4. 定量方法与采集方式结合
如果将上述的定量方法(图2)和质谱数据采集方式(图3)结合起来,就得到了现在基于质谱的蛋白质组学研究的各种策略(图4)。再打个比方,保证吃货们一听就懂:鸡、鱼、肉、蛋、蔬菜要通过炒锅、烤箱、高压锅、微波炉等烹调之后才能变为美食,填饱肚子。同样的,各种定量方法(非标的和标记的)处理的样品,要通过质谱各种采集方式变为电脑中的数据,才能分析并从中得到蛋白的信息。
References
1.A draft map of the human proteome. Nature 509: 575–581 (2014)
2.Mass-spectrometry-based draft of the human proteome. Nature 509: 582–587 (2014)
3.A review: Annu. Rev. Biochem. 80: 273–99 (2011)
4.SILAC: Molecular & Cellular Proteomics 1: 376-386 (2002)
5.iTRAQ: Molecular & Cellular Proteomics 343: 91–99 (2010)
6.SRM: Nature Methods 9: 555–566 (2012)
7.SWATH: Molecular & Cellular Proteomics 11: 1–17 (2012)