无“键”不摧:Science报道低温催化甲烷C-H键活化反应
碳氢(烃类)化合物作为石油化工产业的核心研究对象,除了用作化石燃料成为当今社会的主要能源物质,还可以应用于工业生产在制备其他化学品及聚合材料方面展现多重用途。其中甲烷(CH4)是最简单的碳氢化合物,在地球上储存量巨大。作为天然气的主要成分,它具有热值高、成本低、安全无毒等特点。相比于煤炭、石油等化石燃料,CH4更容易燃烧完全,无烟无尘,仅得到燃烧产物水和二氧化碳。虽然这种碳氢化合物作为清洁能源得到了广泛的利用,但从微观结构上看,CH4的碳原子中心与四个氢原子形成正四面体对称结构,C-H键的平均解离能为ΔdH= 440 kJ/mol,与此同时,CH4还具有较低的质子亲和能(544 kJ/mol),作为Brønsted酸酸性较弱,pKa约为40。由此可见CH4的化学性质相对稳定,C-H键发生均裂与异裂均存在一定困难,在用作起始原料发生进一步转化时受到严重的限制。
尽管如此,人们仍旧希望可以开展有效的方法对CH4进行活化。这一过程不仅可以提供甲醇、甲醛及乙烯等极具工业利用价值的原料,另一方面,将这种可燃气体转化为相应的液态化石燃料虽经历二次化学转化,但可以极大地降低运输成本,从经济角度来看仍旧可以带来可观的收益。使用非均相或均相的金属催化剂实现CH4的活化是目前为止最为行之有效的途径,CH4的C-H键可以通过与催化剂表面的金属中心相互作用降低其活化能,从而发生进一步氧化,但如何控制CH4的选择性催化转化一直是较为严峻的问题。以往大多数催化剂参与的反应中CH4起始发生C-H键断裂为决速步骤,这就导致随后的氧化过程进展迅速难以控制,从而发生复杂的竞争反应。其中一种可行的方法是降低反应温度,理论上可以提高CH4发生氧化的选择性,但目前为止实现其较低温度下发生转化的催化材料尚无报道。
最近,美国佛罗里达大学(UF)的Jason F. Weaver教授团队解决了这一问题,他们在前期大量工作的基础上发展了一种金红石型的Ir金属氧化物作为催化剂,CH4在这种IrO2(110)催化剂的吸附作用下与Ir金属中心形成强作用结合的σ络合物,反应温度低至150 K时便可发生C-H键断裂,进而发生其他转化。研究结果发表在Science 上,第一作者为Zhu Liang 博士。
根据碳氢化合物在催化剂表面的活化原理不同,C-H键的断裂机制可以分为直接解离与前体介导解离两种模式。直接解离是指碳氢化合物分子与催化剂表面在碰撞期间即发生C-H键断裂。而后者则是碳氢化合物分子首先吸附于催化剂表面,并与催化剂相互作用形成前体,随后在适当的条件下发生C-H键断裂,这一催化转化过程相对可控,因而设计CH4在前体介导解离模式下发生氧化的反应选择性更为理想。Jason F. Weaver教授长期从事碳氢化合物催化转化领域的研究工作,他们前期发现后过渡金属氧化物,特别是PdO(101)的特定取向表面可以催化烷烃分子的C-H键发生断裂。该表面存在配位不饱和的金属位点可促进烷烃分子吸附,加强Pd金属中心与烷烃C-H键的相互作用形成σ络合物,氧原子也可作为H原子受体促进C-H键发生解离。他们通过原位测量实验证实了在PdO(101)的作用下,催化剂表面的CH4发生了高效率的氧化。随后根据密度泛函理论(DFT)计算,Jason F. Weaver团队预测CH4在金红石型的RuO2和IrO2表面也可以形成强作用结合的络合物。其中CH4在IrO2(110)表面形成σ络合物的结合能(Ed)比C-H键断裂的活化能(Er)高约40 kJ/mol,当Er小于Ed时,前体介导解离机理的C-H键断裂可以发生,因而他们推测利用该催化剂可能实现低温条件下CH4的活化过程。
作者首先在775 K与氧气分压为5 torr的条件下氧化Ir(100)得到规则的IrO2(110),通过低能电子衍射(LEED)图谱观察到这一金红石型的IrO2(110)晶胞尺寸为3.16 Å × 6.36 Å,晶格矢量沿Ir(100)生长基底的[001]和[110]两个方向排列,配位不饱和的Ir金属原子列被桥连的O原子列分开,相应的Ir金属原子与O原子各缺少一个键合原子形成空配位点。程序升温解吸(TPD)实验定量证实IrO2(110)薄层由46个单原子层组成,厚度约3.5 nm。研究发现CH4在150 K的低温条件下于IrO2(110)表面便可发生C-H键断裂。
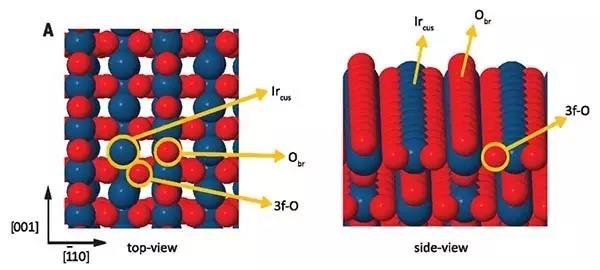
随后他们设计了在88 K条件下分别通过程序升温反应谱(TPRS)测定不同CH4覆盖率下CH4的反应情况,TPRS曲线表明吸附于IrO2(110)表面的大部分CH4都被氧化为CO、CO2和H2O,继续升高温度可以使产物发生解吸。他们还通过实验数据证实了程序升温期间,大量CH4与IrO2(110)表面配位不饱和的Ir金属中心以及桥连的O原子作用,CH4在IrO2(110)表面经历了C-H键活化过程。
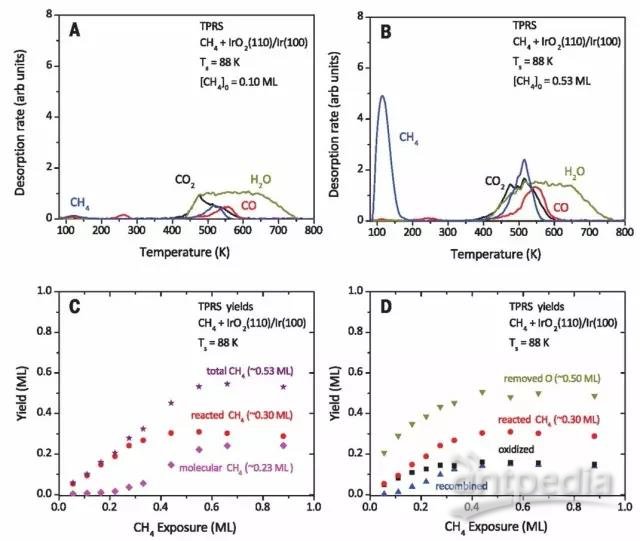
图2. IrO2(110)表面CH4的吸附及反应
他们还利用CH4发生催化转化的前体介导解离动力学模型推导出CH4发生C-H键断裂的概率与催化剂表面温度的关系,最终给出以下Arrhenius方程。其中νj和Ej分别表示反应的指前因子和活化能,Ts为催化表面的温度,ξ为CH4分子的解吸概率,S0为CH4分子覆盖率趋近于零时C-H键断裂的概率(起始解离概率)。
接下来他们通过设计不同催化表面温度的TPRS实验,以得到的实验数据拟合工作曲线,计算出不同催化表面温度下CH4的起始解离概率,最终通过这些数据计算出Arrhenius方程中的各个参数,从而估算出低温条件下CH4在低覆盖率的IrO2(110)表面形成σ络合物的结合能(Ed)约为38 kJ/mol,C-H键断裂的活化能(Er)为28.5 kJ/mol。为了便于对IrO2(110)的催化活性进行评测,作者利用同样的方法估算出PdO(101)催化CH4发生C-H键断裂的活化能约为56 kJ/mol,由此发现,低温条件下IrO2(110)对促进CH4发生高选择性氧化具有十分优异的效果。
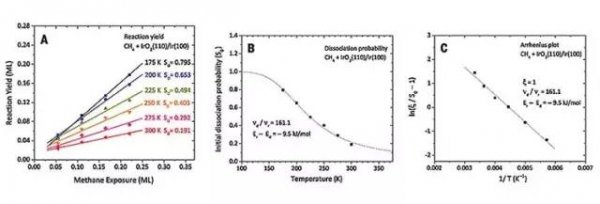
图4. IrO2(110)表面CH4发生C-H键活化的动力学分析
总结
Jason F. Weaver教授报道的金红石型IrO2(110)催化剂对有效实现CH4的选择性官能团化具有十分重要的指导意义,但目前为止,使用该催化剂仅可以实现CH4向高氧化态的CO、CO2转化。人们可以借鉴这一催化模型寻找更为理想的金属催化剂,例如修饰IrO2(110)催化剂的表面对其进行合理的改性,限制其氧化能力,或结合其他材料使CH4在发生C-H键活化后与特定的共反应物作用,得到丰富多样的高附加值产物。