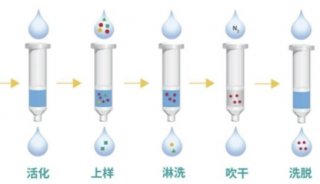
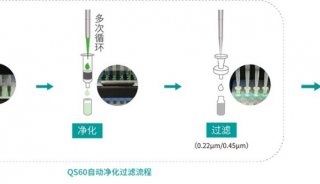
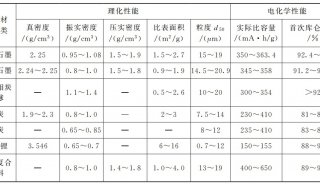
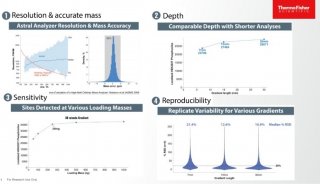
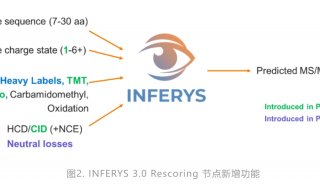
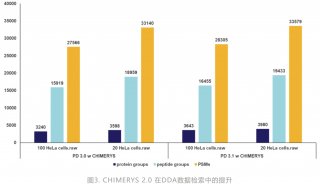
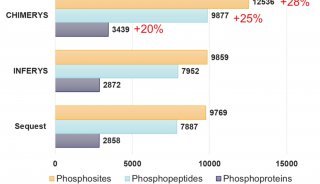
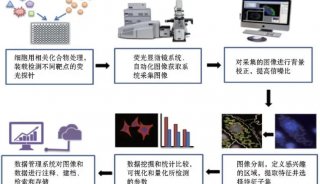
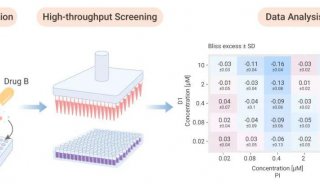
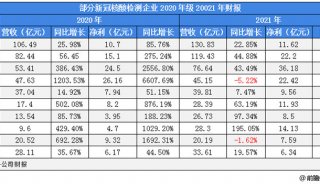
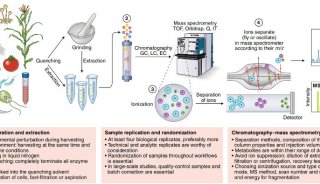
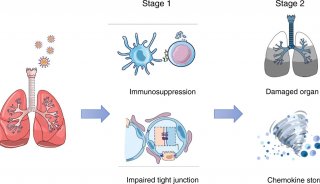
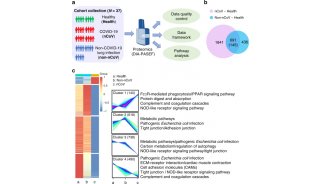
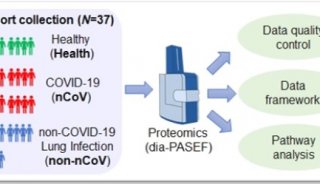
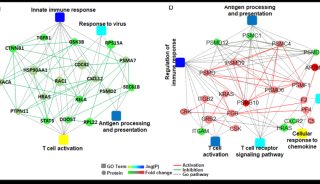
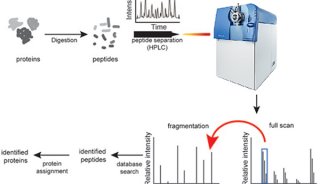
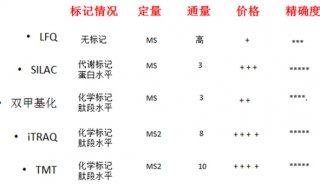
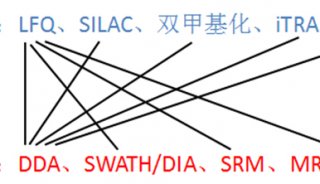
Proteome and Phosphoproteome Quantification by DIA on Orbitrap (一)
Overview
1) Purpose: Data Independent Acquisition (DIA) has been applied for large-scale protein and PTM quantification on the Thermo Scientific ™ Q Exactive HF and Orbitrap Fusion ™ Tribrid ™ instruments, to evaluate performance of the ultra-high field Orbitrap ™ based DIA strategy for complicated sample
analysis.
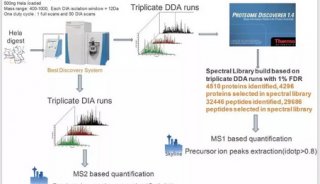
2) Methods: HeLa protein digest and enriched rat phosphorylated protein digest were analyzed on a Q Exactive HF MS and a Orbitrap Fusion MS by DDA and DIA methods, respectively. Data processing was performed on Skyline (version 3.1) using DDA database searching result (Proteome Discoverer version 1.4) as spectra library.
3) Results: A total of 21710 peptides corresponding to 3597 protein groups were extracted with high confidence (Q value ≤ 0.01) as well as good reproducibility from only 200 ng HeLa protein digest within 2-hour LC gradient. Moreover, A total of 9990 phosphorylated peptides were characterized and quantified with the same criterion. The results indicate thestrengths of DIA on ultra high-field Orbitrap MS.
Introduction
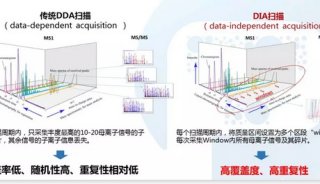
Orbitrap platforms based Data Independent Acquisition (DIA) is becoming increasingly popular, owing to its high sensitivity, selectivity and reproducibility compared to classic Data Dependent Acquisition (DDA) as well as other DIA techniques.
Meanwhile, the flexibility of Orbitrap MS permits the development of novel acquisition strategies as well as solving current puzzles in proteomics such as PTM quantification.
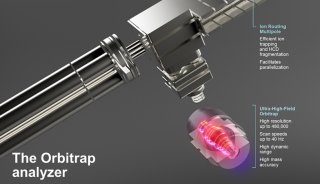
The latest ultra-high field Orbitrap technology combines the smaller size Orbitrap cell (20 mm ID) with 5 kV of the central electrode voltage, and realizes 1.8-fold scan rate increase at the same resolution or 1.8-fold resolution increase at the same frequency. State-of-the-Art performance and flexibility are thus accessible on ultra-high field Orbitrap based Q Exactive HF and Orbitrap Fusion mass spectrometers.
Herein, we utilized the ultra-high field Orbitrap MS for largescale proteomic and phosphoproteomic DIA analysis, to evaluate performance and strengths of the ultra-high field Orbitrap based DIA strategy. The strategy shows outstanding quantification ability compare to current DIA techniques and provides a powerful tool for high-throughput proteomic sample application.
Methods
1) Materials
Commercial HeLa protein digest was purchased from Pierce ™ (PN: 88328), with a loading amount of 200 ng for each run. Phosphoprotein sample was enriched from rat tissue and digested by trypsin, with a final loading amount of 700 ng for each run.
2) Liquid Chromatography
Each sample was analyzed on a Thermo Scientific ™ EASYnLC ™ 1000 nanoflow LC coupled to a Thermo Scientific ™ Nanospray Flex ™ ion source. The sample was loaded onto a nano-C18 column and separated at a flow rate of 300 nL/min with following gradient: 0–3 min, 3–7% buffer B (0.1% fomic
acid in acetonitrile); 3–95 min, 7–22% B; 95–113 min, 22–35% B; 113–116 min, 35–90% B; 116–120 min, 90% B.
3) Mass Spectrometry
DDA performed 120K resolution MS scan and then triggered top 15 precursors (QE HF) or acquired under 3-second topspeed mode for 60K resolution MS/MS scans. The MS/MS AGC target value was set at 1e6 (QE HF) or 5e4 (OT Fusion) with 100-110 ms of max injection time (Figure 1A).
DIA collected high-resolution MS/MS spectra by sequential 25 amu step length (26 amu isolation window, 0.5 amu overlapping between windows) from m/z 400 to 1200 for HCD fragmentation (28%). MS/MS ions were detected using 30K/60K of Orbitrap resolution, 1e6 (QE HF) or 1e5 (OT Fusion)
of AGC target value and 85–100 ms of max injection time. MS scan was also performed before each DIA cycle (Figure 1B).
4) Data Analysis
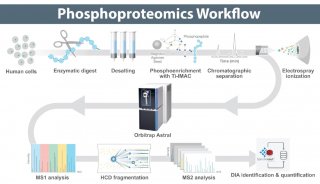
Database searching was performed on Thermo Scientific ™ Proteome Discoverer ™ software (version 1.4) with Uniprot human or rat protein database. Mass tolerance of precursor and fragment were set at 10 ppm and 0.02 Da, respectively. Results were validated by Percolator with q < 0.01 and then used as spectral library for DIA analysis.
DIA Data processing was performed on Skyline software (version 3.1). Three precursor (mono) isotopes and top six fragment ions in spectral library of each peptide were selected as transitions for peak extraction. Results were refined by mProphet using decoy sequences and then validated by Q value (< 0.01).
Results
1) Data Independent Acquisition for Large- Scale HeLa Proteome Analysis
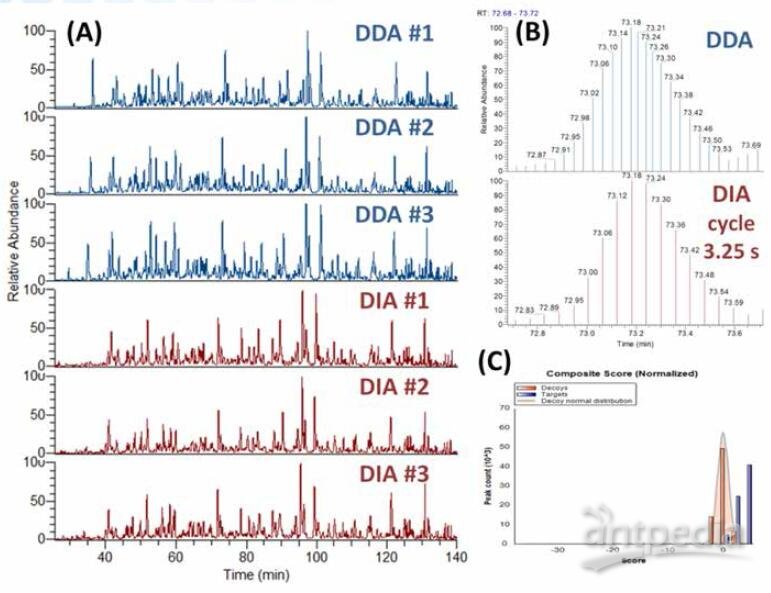
Three replicates were performed for each DDA and DIA methods, with a loading amount of 200 ng Hela digest for each run. DDA and DIA were proceeded under identical sample and LC conditions to evaluate and compare results of the two strategy directly.
Figure 2 shows MS1 chromatograms of the DDA and DIA replicates. DIA limits cycle time at 3–3.5 seconds and thus maximizes sensitivity and accuracy by increasing AGC value, maximum injection time and resolution within the cycle time limitation. The zoomed-in chromatograms display scan point distributions of an example DDA and DIA peak. The cycle time of DIA is fixed and thus the chromatogram scan points are uniformly distributed, compared to that of DDA data.
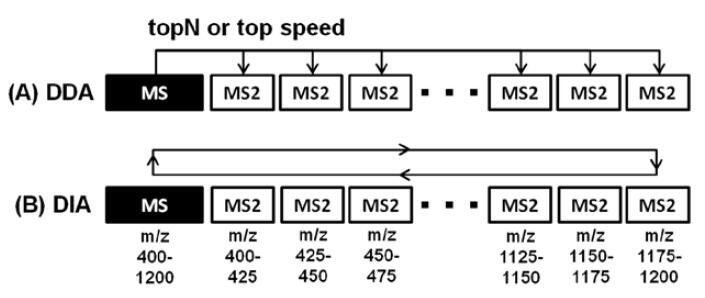
FIGURE 1. Schematic comparison of data dependent (A) and data independent (B) acquisition methods on ultra-high field Orbitrap platforms.
-
并购

-
焦点事件

-
产品技术
-
会议会展

-
焦点事件

-
企业风采

-
企业风采

-
企业风采

-
企业风采

-
企业风采

-
企业风采

-
焦点事件

-
焦点事件

-
焦点事件
-
会议会展

-
企业风采

-
焦点事件

-
市场商机

-
焦点事件
-
产品技术
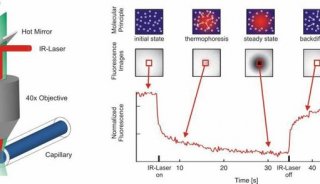
-
焦点事件

-
企业风采

-
产品技术

-
企业风采

-
焦点事件

-
焦点事件
-
焦点事件
-
产品技术

-
科技前沿

-
企业风采

-
焦点事件

-
焦点事件

-
精英视角

-
焦点事件

-
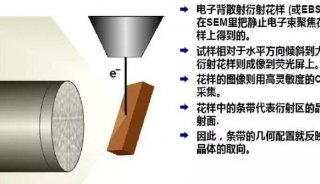
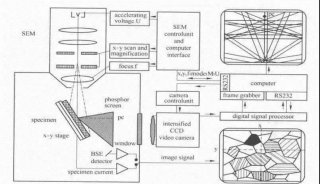
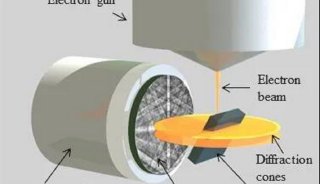
技术原理
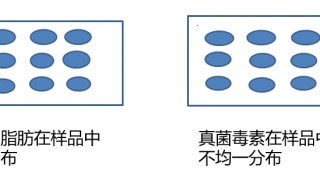
-
产品技术

-
焦点事件
-
综述

-
焦点事件

-
综述

-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件

-
企业风采

-
企业风采

-
企业风采

-
焦点事件
-
焦点事件
-
焦点事件
-
焦点事件

-
焦点事件
-
焦点事件
-
焦点事件

-
产品技术

-
企业风采

-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件
-
焦点事件
-
企业风采

-
企业风采

-
企业风采

-
企业风采

-
焦点事件
-
焦点事件
-
企业风采

-
企业风采

-
企业风采

-
综述
-
科技前沿
-
焦点事件

-
产品技术