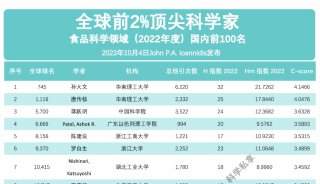
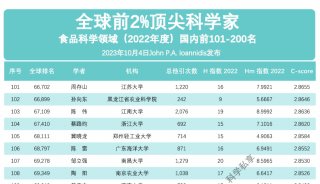
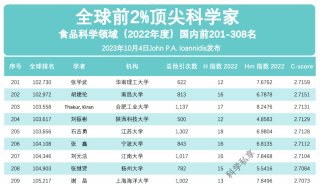
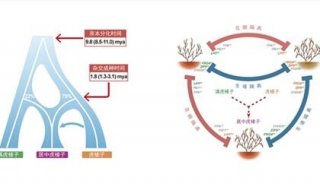
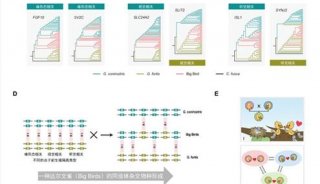
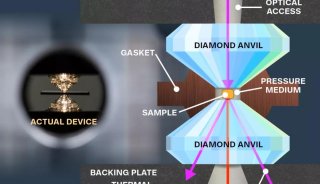
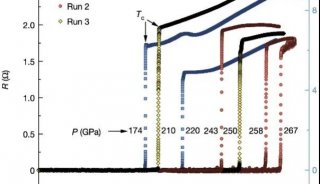
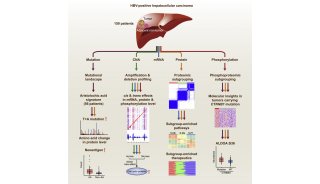
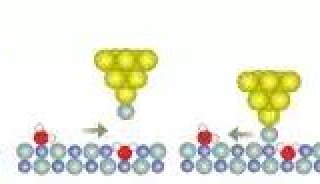
科学家利用基因组编辑扩大昆虫学研究
基因组测序,即科学家使用实验室方法来确定特定生物体的基因组成,正在成为昆虫研究的一种常见做法。对昆虫生物学的深入了解有助于科学家更好地管理昆虫,包括那些对生态系统有益的昆虫,以及那些破坏食物供应并通过携带疾病威胁人类健康的昆虫。
研究人员开发了一种名为fanflow4insect的工作流方法,可以注释昆虫的基因功能。在功能注释中,科学家收集有关基因生物学特性的信息。该团队的新方法使用转录序列信息以及基因组和蛋白质序列数据库。通过fanflow4insect,研究小组对日本竹节虫和家蚕的功能信息进行了标注,包括基因表达和序列分析。他们的工作流程提供的功能性注释信息将极大地拓展昆虫学研究使用基因组编辑的可能性。
这个团队与来自广岛大学、东京农业技术大学和日本理研综合医学科学中心的科学家一起,于6月27日在《昆虫》杂志上发表了他们的fanflow4insect方法。
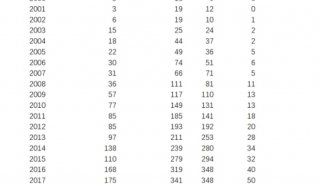
昆虫是如此的多样化和丰富,以至于科学家需要一种大规模研究它们的方法。这促使科学家们开始对昆虫的基因组进行测序。截至2022年5月,科学家已经破译并登记了约3000种昆虫的基因组。他们还利用长读测序技术进一步加快昆虫基因组测序的步伐。
下一代测序技术使研究人员更容易解码大量昆虫的基因组和它们的转录序列。然而,这些序列的生物学解释仍然是转录组分析的主要瓶颈。转录组是生物体RNA分子的总和。转录组分析是功能注释的重要第一步,是选择基因组编辑靶点的重要线索。
因为一些昆虫的基因组比人类的基因组还大,所以整个基因组测序的困难过程就更加复杂了。因此,科学家们正在利用下一代测序技术(也被称为RNA测序)进行转录组测序,作为评估大型基因组大小昆虫的工具。有了这个强大的工具,科学家们可以通过组装数千万个reads,有效地识别出一个特定组织中数万个可能的基因。然后他们将基因序列组装成转录单位进行鉴定。但这种类型的分析依赖于科学家能够访问全面的数据集及其功能注释。数据库确实存在,但它们无法跟上昆虫基因组测序的增长速度。
随着转录组分析变得越来越流行,许多研究小组都在运行自己的研究管道,从各种研究中获得的转录单位的信息都在逐项研究的基础上进行报告。这些管道是用于处理基因组测序数据的算法集。但科学家们需要一种方法,将所有从事这类研究的不同小组的功能性注释整合到公共数据库中。
在目前的研究中,研究团队使用他们新开发的fanflow4insect为家蚕创建了一个功能性注释管道。然后,研究人员还测试了fanflow4insect日本竹节虫的转录组。一旦目标生物的基因组或转录组被解码,功能注释是加速目标基因选择的最重要过程之一。该论文的第一作者和通讯作者、广岛大学生命综合科学研究生院教授hideasa Bono表示:“fanflow4insect工作流程获得的功能性注释信息将极大地拓展利用基因组编辑技术进行昆虫学研究的可能性。”
昆虫的fanflow4insect工作流已经在GitHub上公开开发,可以免费访问。结合由表达衍生的功能注释,fanflow4insect的数据可用于不同表型昆虫的比较研究。“使用fanflow4insect,我们将注释产生有用物质的昆虫。这项研究的最终目标是利用计算机模拟设计昆虫的分子网络,”Bono说。
-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件

-
会议会展

-
焦点事件

-
焦点事件

-
项目成果
-
焦点事件

-
会议会展

-
焦点事件

-
项目成果
-
焦点事件

-
会议会展

-
焦点事件
-
企业风采

-
科技前沿

-
科技前沿

-
焦点事件

-
焦点事件

-
科技前沿

-
焦点事件

-
焦点事件

-
焦点事件

-
科技前沿

-
焦点事件

-
科技前沿

-
科技前沿

-
科技前沿

-
焦点事件

-
科技前沿

-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件

-
焦点事件

-
会议会展

-
企业风采

-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件
-
精英视角

-
企业风采

-
项目成果

-
焦点事件
-
企业风采

-
焦点事件

-
焦点事件

-
科技前沿

-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件

-
科技前沿

-
科技前沿
-
科技前沿
-
科技前沿

-
科技前沿
-
科技前沿

-
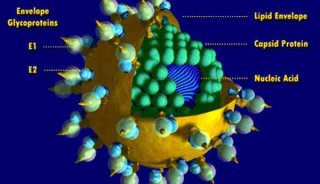
科技前沿
-
焦点事件

-
科技前沿

-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件

-
科技前沿
-
科技前沿

-
焦点事件

-
项目成果
-
产品技术

-
科技前沿

-
焦点事件

-
会议会展

-
焦点事件
-
焦点事件

-
项目成果
-
焦点事件
-
焦点事件
-
项目成果

-
焦点事件

-
焦点事件

-
企业风采

-
焦点事件

-
焦点事件

-
企业风采

-
企业风采

-
科技前沿
-
焦点事件

-
焦点事件

-
焦点事件

-
科技前沿
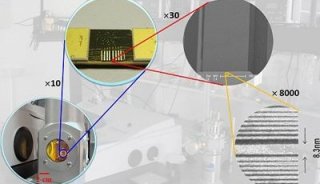
-
焦点事件
-
焦点事件
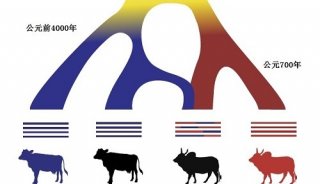
-
焦点事件
-
焦点事件

-
项目成果

-
项目成果
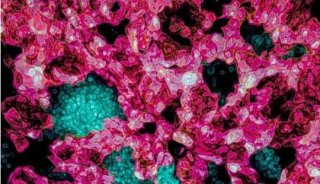
-
焦点事件
-
焦点事件
-
焦点事件

-
焦点事件

-
实验室动态

-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件
-
焦点事件

-
焦点事件
-
项目成果

-
项目成果
-
项目成果

-
焦点事件
-
焦点事件
-
项目成果

-
项目成果

-
焦点事件

-
企业风采

-
焦点事件

-
焦点事件
-
企业风采

-
焦点事件

-
科技前沿

-
焦点事件
-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件

-
精英视角

-
焦点事件
-
焦点事件

-
焦点事件

-
企业风采

-
焦点事件

-
焦点事件

-
项目成果
-
焦点事件
-
焦点事件
-
焦点事件
-
科技前沿

-
焦点事件

-
项目成果
-
焦点事件

-
焦点事件

-
项目成果

-
会议会展

-
焦点事件

-
焦点事件
-
会议会展

-
焦点事件
-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件
-
焦点事件
-
焦点事件
-
焦点事件

-
实验室动态

-
会议会展

-
焦点事件
-
焦点事件