分子光谱分析法第三弹—红外光谱
红外光谱(infrared absorption spectrum ,IR)又称分子振动转动光谱,属分子吸收光谱。样品受到频率连续变化的红外光照射时,分子吸收其中一些频率的辐射, 使振-转能级从基态跃迁到激发态,相应于这些区域的透射光强减弱,记录百分透过率T%对波数或波长的曲线,即红外光谱。
红外光谱的研究开始于20世纪初期,自1940年商品红外光谱仪问世以来,红外光谱在有机化学研究中得到广泛的应用。近几十年来一些新技术(如发射光谱、光声光谱、色—红联用等)的出现,使红外光谱技术得到更加蓬勃的发展。
红外光去的划分
通常将红外光谱分为三个区域:近红外区(0.75~2.5μm)、中红外区(2.5~25μm)和远红外区(25~300μm)。一般说来,近红外光谱是由分子的倍频、合频产生的;中红外光谱属于分子的基频振动光谱;远红外光谱则属于分子的转动光谱和某些基团的振动光谱。由于绝大多数有机物和无机物的基频吸收带都出现在中红外区,因此中红外区是研究和应用最多的区域,积累的资料也最多,仪器技术最为成熟。通常所说的红外光谱即指中红外光谱。

红外光谱分析原理
将一束不同波长的红外射线照射到物质的分子上,某些特定波长的红外射线被吸收,形成这一分子的红外吸收光谱。每种分子都有由其组成和结构决定的独有的红外吸收光谱,据此可以对分子进行结构分析和鉴定。
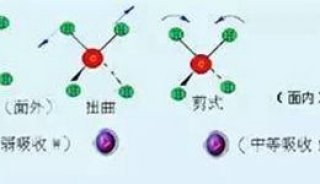
分子的振动形式
分子的振动分为伸缩振动和变形振动两类。伸缩振动是沿原子核之间的轴线作振动,键长有变化而键角不变,用字母υ来表示。
伸缩振动分为不对称伸缩振动υas和对称伸缩振动υs。
变形振动是键长不变而键角改变的振动方式,用字母δ表示。
红外吸收光谱产生的条件
满足两个条件:
(1)辐射应具有能满足物质产生振动跃迁所需的能量
(2)辐射与物质间有相互偶合作用。
对称分子:没有偶极矩,辐射不能引起共振,无红外活性。如:N2、O2、Cl2 等。
非对称分子:有偶极矩,红外活性。
红外光谱仪的组成
组成:光源、单色器、样品池、检测器。
光源:①能提供连续的辐射;②光强度足够大;③在整个光谱区内光谱强度不随波长有明显变化;④光谱范围宽;⑤使用寿命长,价格低。 钨灯——可见光区 320~2500nm,氢灯或氘灯——紫外光区 195-375nm,U3010(碘钨灯、氘灯)波长范围190-900nm。
单色器:包括狭缝、准直镜、色散元件。
吸收池:玻璃—由于吸收紫外UV光,仅适用于可见光区; 石英—适用于紫外和可见光区。
检测器(将光信号转变为电信号的装置):光电管;光电倍增管;二极管阵列检测器。
记录装置:讯号处理和显示系统。
红外光谱法对样品要求
样品要求以及注意事项:参照池和吸收池应是一对经校正好的匹配的吸收池,材料和规格一致;使用前后应将洗手池洗净,测量时不能用手接触窗口;
已匹配好的比色皿不能用炉子和火焰干燥,不能加热,以免引起光程长度上的改变;
选择适宜波长的入射光,由于有色物质对光有选择性吸收,为了使测定结果有较高的灵敏度,必须选择溶液最大吸收波长的入射光;
控制吸光度A的准确的读数范围,由朗伯-比耳定律可知,吸光度只有控制在0.2--0.7读数范围内时,测量的准确度较高;
选择参比溶液,参比溶液是用来调节仪器工作零点的。若样品溶液,试剂,显色剂无色可用蒸馏水作参比溶液,反之应采用不加显色剂的样品液作参比溶液。
红外吸收光谱的特点
1.只有振-转跃迁,能量低
2.应用范围广,除单原子分子及单核分子外,几乎所有物质均有红外吸收;
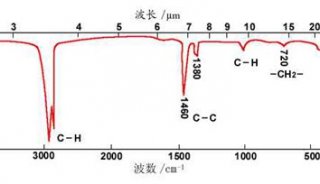
3.分子结构更为精细的表征,通过IR谱的波数位置、波峰数目具强度确定分子基团、分子结构。
4.可以进行定量分析
5.样品不限形式,用量少,不破坏样品
6.分析速度快
7.可与色谱等联用具有强大的定性功能。
红外光谱法的应用
一、定性分析
1. 已知物的鉴定
将试样谱图与标准谱图对照或与相关文献上的谱图对照。
2. 未知物结构分析
如果化合物不是新物质,可将其红外谱图与标准谱图对照;
如果化合物为新物质,则须进行光谱解析,其步骤为:
(1)该化合物的信息收集:试样来源、熔点、沸点、折光率、旋光度等;
(2)不饱和度的计算:
通过元素分析得到该化合物的分子式,并求出其不饱和度W。
W=0 时,分子是饱和的,分子为链状烷烃或其不含双键的衍生物;
W=1 时,分子可能有一个双键或脂环;
W=2 时,分子可能有一个三键或两个双键;
W=4 时,分子可能有一个苯环。
注意:一些杂原子如S、O不参加计算。
(3)查找基团频率,推测分子可能的基团;
(4)查找红外指纹区,进一步验证基团的相关峰;
(5)能过其它定性方法进一步确证:UV-Vis、MS、NMR、Raman光谱等。
二、定量分析
红外光谱的谱带较多,选择余地大,所以能方便地对单一组分或多组分进行定量分析,并且该方法不受试样状态的限制。但红外光谱法的灵敏度较低,尚不适于微量组分测定。
定量分析的依据也是基于朗伯-比尔定律,通过对特征吸收谱带强度的测量求组分含量。
但红外谱图复杂,相邻峰重叠多,难以找到合适的检测峰;红外谱图峰形窄,光源强度低,检测器灵敏度低,测定时必须使用较宽的狭缝,从而导致对朗伯-比尔定律的偏离;红外测定时吸收池厚度不易确定,利用参比难以消除吸收池、溶剂的影响。
红外光谱仪的使用注意事项
1.测定时实验室的温度应在15~30℃,相对湿度应在65%以下,所用电源应配备有稳压装置和接地线。因要严格控制室内的相对湿度,因此红外实验室的面积不要太大,能放得下必须的仪器设备即可,但室内一定要有除湿装置。
2.如所用的是单光朿型傅里叶红外分光光度计(目前应用最多),实验室里的CO2含量不能太高,因此实验室里的人数应尽量少,无关人员最好不要进入,还要注意适当通风换气。
3.如供试品为盐酸盐,因考虑到在压片过程中可能出现的离子交换现象,标准规定用氯化钾(也同溴化钾一样预处理后使用)代替溴化钾进行压片,但也可比较氯化钾压片和溴化钾压片后测得的光谱,如二者没有区别,则可使用溴化钾进行压片。
4.为防止仪器受潮而影响使用寿命,红外实验室应经常保持干燥,即使仪器不用,也应每周开机至少两次,每次半天,同时开除湿机除湿。特别是霉雨季节,最好是能每天开除湿机。
5.红外光谱测定最常用的试样制备方法是溴化钾(KBr)压片法(药典收载品种90%以上用此法),因此为减少对测定的影响,所用KBr最好应为光学试剂级,至少也要分析纯级。使用前应适当研细(200目以下),并在120℃以上烘4小时以上后置干燥器中备用。如发现结块,则应重新干燥。制备好的空KBr片应透明,与空气相比,透光率应在75%以上。
6.压片法时取用的供试品量一般为1~2mg,因不可能用天平称量后加入,并且每种样品的对红外光的吸收程度不一致,故常凭经验取用。一般要求所没得的光谱图中绝大多数吸收峰处于10%~80%透光率范围在内。最强吸收峰的透光率如太大(如大于30%),则说明取样量太少;相反,如最强吸收峰为接近透光率为0%,且为平头峰,则说明取样量太多,此时均应调整取样量后重新测定。
7.测定用样品应干燥,否则应在研细后置红外灯下烘几分钟使干燥。试样研好并具在模具中装好后,应与真空泵相连后抽真空至少2分钟,以使试样中的水分进一步被抽走,然后再加压到0.8~1GPa(8~10T/cm2)后维持2~5min。不抽真空将影响片子的透明度。
8.压片时KBr的取用量一般为200mg左右(也是凭经验),应根据制片后的片子厚度来控制KBr的量,一般片子厚度应在0.5mm以下,厚度大于0.5mm时,常可在光谱上观察到干涉条纹,对供试品光谱产生干扰。
9.压片时,应先取供试品研细后再加入KBr再次研细研匀,这样比较容易混匀。研磨所用的应为玛瑙研钵,因玻璃研钵内表面比较粗糙,易粘附样品。研磨时应按同一方向(顺时针或逆时针)均匀用力,如不按同一方向研磨,有可能在研磨过程中使供试品产生转晶,从而影响测定结果。
研磨力度不用太大,研磨到试样中不再有肉眼可见的小粒子即可。试样研好后,应通过一小的漏斗倒入到压片模具中(因模具口较小,直接倒入较难),并尽量把试样铺均匀,否则压片后试样少的地方的透明度要比试样多的地方的低,并因此对测定产生影响。另外,如压好的片子上出现不透明的小白点,则说明研好的试样中有未研细的小粒子,应重新压片。
10.压片用模具用后应立即把各部分擦干净,必要时用水清洗干净并擦干,置干燥器中保存,以免锈蚀。