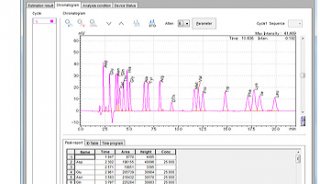
测序结果的分析
测序都是从5'端进行的,正向和反向测序是指对DNA的两条互补链分别测序,通常两个方向测序结果经校读后完全一致才能认为得到可靠结果。生工测序结果一般都提供两个文档,一个是TEXT的序列文档,一个是用Chromas软件打开的ABI文档。
1.寻找引物
http://blast.ncbi.nlm.nih.gov/Blast.cgi 比对,去除引物序列,找到目的片段。
在DNAMan上进行比对,看引物能不能比对上(一个不变,一个反向互补),如果比不上,那可能就不是你要的序列,如果能比上,上游以引物第一个为分界线,去除前面的;下有一最后一个为分界线,去除后面的,剩下的就是目的序列。然后在NCBI上Blast.就OK了。
批注:PCR产物进行测序的结果可能不包含引物序列
2.将找到的对应目的片段转成*.txt格式
3.下载BioEdit软件
第一:打开Bioedit软件,导入拼接好的样品序列与标准亚型参考序列
File—New Alignment—Sequence—New Sequence—导入拼接好的样品序列和标准参考序列(从TEXT文档利用复制粘贴工具)—Apply and close —保存结果—关闭窗口
第二:点击菜单栏上按钮“Accessory Application”,选择“Clustalw Multiple Alignment”
File—Open—Accessory Application—Clustalw Multiple Alignment
第三:比对结束后,删除比对序列两端的多余序列,使所有序列等长
选择需要编辑的序列—Sequence—Edit Sequence—进行序列的编辑—保存修改后结果
第四:选择“Sequence”菜单下的“Gaps”,点击“Lock Gaps”
第五:将比对后的序列保存为Fasta 格式文档
4.下载MAGE4.0软件
1) 打开MEGA软件,选择“File”菜单栏中的“Convert To MEGA Format”,把序列文件的格式转换为meg文档保存;
2) 双击序列的meg文档,选择“Nucleotide Sequences”,点击“OK”;
3) 程序运行中询问是否为蛋白编码序列,选择“NO”;
4) 在MEGA操作界面选择“Phylogeny”菜单栏下“Bootstrap Test of Phylogeny”中的“Neibour-Joining”;
5) 选择“Test of Phylogeny”栏中的“Bootsrap”,“Replications”设定为1 000;在“Options Summary”栏中的“Model”项中,设定参数为“Kimura 2-Paramete”r,最后选择“Compute”;
6) 将分析结果采用Los Alamos HIV序列库提供的HIV-BLAST和Subtyping工具进行验证