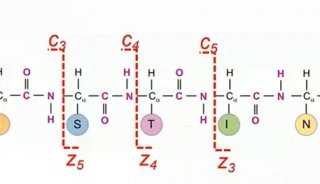
通过软件创新打造更强大的4D-蛋白质组学技术

在过去的二十年中,新技术、新方法的重大进步使蛋白质组学成为蛋白质科学家、生物学家和临床研究人员的一个极其强大的工具1。随着分析仪器的不断发展,蛋白质组学研究的每一项技术进步都会产生更多的数据。与此同时也给生物信息学软件的开发带来了新挑战。
当代基于高通量质谱的蛋白质组学方法使人们对生命过程获得更深入的了解,同时产生大量的数据。这些原始数据需要高效自动化的计算机算法来保障蛋白质的定性定量结果的可靠性。
全球许多实验室正在使用MaxQuant处理蛋白质组学数据,并借助精确蛋白质和肽定量算法获得可靠结果。MaxQuant由来自德国马克斯·普朗克生物化学研究所(MPIB)的计算系统生物化学组开发,免费提供给学术和非学术研究人员。该小组还开发了Perseus软件平台,用于解释蛋白质定量和相互作用数据以及翻译后修饰(PTMs)数据。
MaxQuant拥有强大的算法,可进行全面的数据分析。它使用肽搜索引擎Andromeda,并与Perseus结合在一起,为下游生物信息学分析提供了完整的解决方案2。MaxQuant通过标记对样品进行定量、通过MaxLFQ算法对非标记的数据进行定量,凭借先进的非线性再校准算法获得了较高的肽质量准确性。
MaxQuant用于4D-蛋白质组学
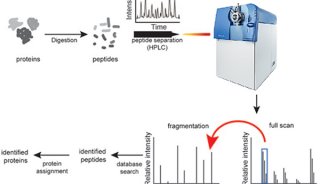
MaxQuant通常用于液相色谱和串联质谱(LC-MS/MS)鸟枪法蛋白质组学(shotgun proteomics)。这是一种识别复杂混合物中蛋白质的方法,用以提供更广泛的动态范围和蛋白质覆盖范围。
鸟枪法蛋白质组学是基于MS最常用的方法,通过在MS分析之前将蛋白质消化成肽段来研究蛋白质。
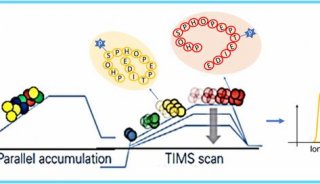
离子淌度可以为基于LC-MS的鸟枪蛋白质组学增加一个新的维度,成为提高蛋白质组的覆盖率、定量精度和动态范围的潜在推动力。但是新增加的淌度信息需要合适的软件来提取由m/z、保留时间、离子淌度和信号强度共同构建的四维(4D)数据。增加离子淌度信息对蛋白质组学领域的影响显而易见。例如被广泛应用于蛋白质组学研究的,布鲁克的timsTOF Pro,提供了更高的灵敏度、选择性和MS/MS采集速度。新颖的设计允许离子在前段累积的同时后段的离子则根据其离子淌度依次释放,在随后的扫描中,选定的前体可以作为MS/MS的目标。这个过程称为平行累积连续碎列,或PASEF®3。
独特的捕集离子淌度(TIMS)技术帮助研究人员获得所有检测离子的碰撞截面(CCS)值,且具有高度可重复性的这些CCS值可用于进一步提高系统的选择性,从复杂样品和短梯度分析中获得更加可靠的相对定量信息。
相关研究表明:结合了TIMS技术和PASEF方法的LC-MS鸟枪蛋白质组学,如虎添翼,在提高蛋白质组的覆盖率、定量精度和动态范围层面拥有无限潜能,从而实现快速超灵敏分析。PASEF技术可以提高分析速度,在较短的时间内分析更多的样本,但同时也产生了大量的谱图数据。如何处理大量的样本群成为一个棘手的难题。

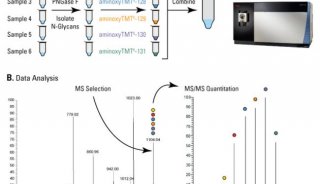
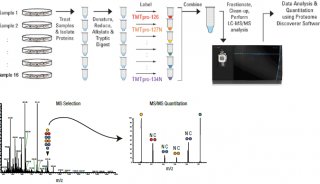
图1:MaxQuant软件全面支持布鲁克4D-蛋白质组学数据解析
MPIB软件开发人员采用MaxQuant shotgun蛋白质组学工作流程,从timsTOF Pro数据中提取丰富的信息,掌控由保留时间、离子淌度、质量和信号强度这些特征所构建的4D空间,使其更有利于肽、蛋白质和PTM的识别和定量。
TIMS对于软件开发人员而言具有挑战性,因为它不是新增添一个层面的信息,而是增加了一个新的维度。新的的MaxQuant 4D-Proteomics工作流程可以处理通过PASEF,dia-PASEF和Mobility Offset Mass Aligned(MOMA)生成的数据。
增加新维度可以延长算法处理时间,这对4D-蛋白质组学的软件开发提出了重大挑战。 最终,通过优化MaxQuant中的计算时间克服了这一问题,保证用户可以在合理的时间范围内获得良好的结果。
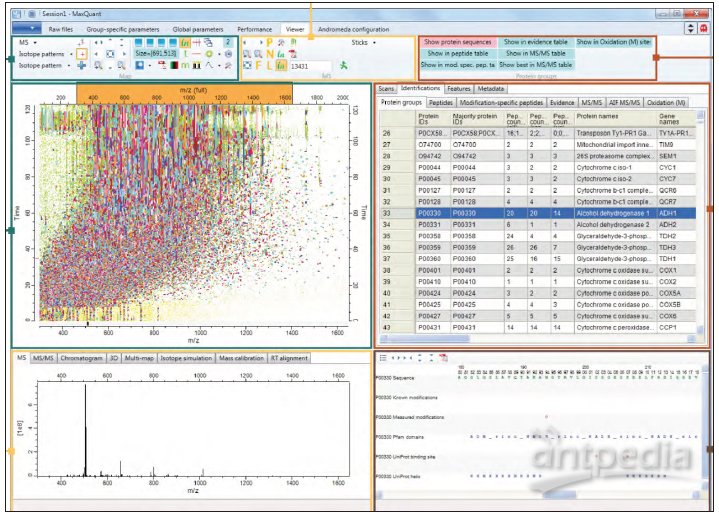
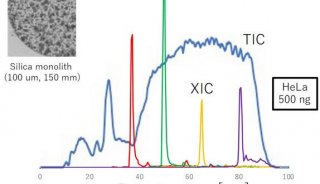
图2:用于分析大型质谱数据集的MaxQuant定量蛋白质组学软件包的屏幕截图。资料来源:Maxquant.org
4D-蛋白质组学的新应用
随着质谱技术的发展,MaxQuant的应用范围不断扩大,性能不断提升以满足蛋白质组学领域的未来需求。这些改进为肽段、蛋白和PTM的定性定量分析中提供更高的灵敏度和选择性,从而帮助科学家开发蛋白质组学的新方法与新应用。仪器的升级也促进了MaxQuant的持续发展。MPIB软件开发团队的目标始终是扩展和改进MaxQuant,以满足生物过程的复杂性和日益更新的质谱技术。
01 蛋白质组学临床研究
MPIB计算系统生物化学团队认为,蛋白质组学临床研究将是未来4D-蛋白质组学的主要应用之一。目前,他们正与多个临床小组合作,将基于MS的蛋白质组学引入临床实践。然而,对来自病人样本的蛋白质组学数据的分析需要特殊算法,所需解决的问题包括:如何从个体差异性大的数据中提取有意义的蛋白质表达特征、如何将患者的基因组背景整合到蛋白质组学数据分析以及如何确定生物标志物并合理推测其效用。
为了解决上述问题,MPIB软件开发人员正在努力利用机器学习算法对患者进行分类,并采用特征选择算法来提取预测性蛋白质特征。不过新增的临床测试引擎为软件带来了挑战,目前尚不清楚蛋白质组学是否可以指导观察分子或可以成为临床诊断的主要组成部分。
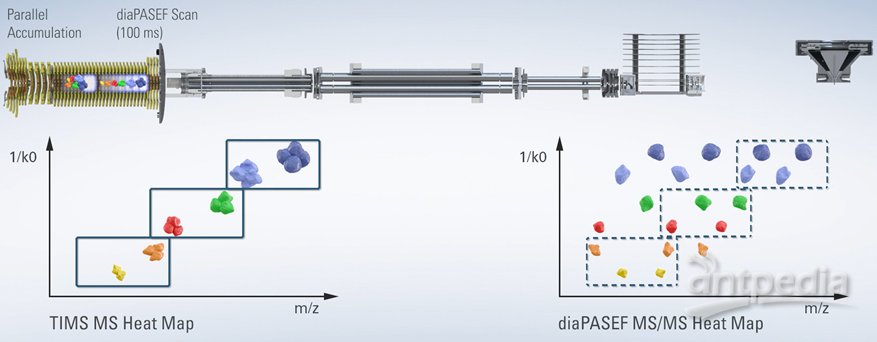
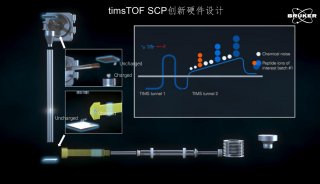
图3:timsTOF Pro仪器的设计。资料来源:www.Bruker.com
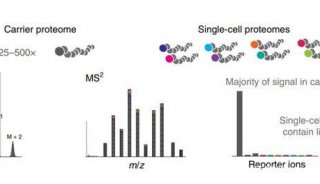
02 单细胞蛋白质组学
当今实验室从单细胞基因组学和单细胞转录组学研究中获得了大量的数据集,而单细胞蛋白质组学(SC-蛋白质组学)则是新兴领域。单细胞蛋白质组学使研究人员能够对单个细胞中的蛋白质进行定性定量分析,从而避免了从细胞信使RNA水平推断蛋白质的需要4。不过这也给计算分析带来了新的挑战。
伴随蛋白质组学的发展,MPIB研究人员对未来的需求充满期待,并密切关注新兴技术以建立量化标准。开发者们认为单细胞蛋白质组学将在未来几年内得以实现。实现这一目的关键在于提高仪器的灵敏度和相应操作软件的扩展更新。
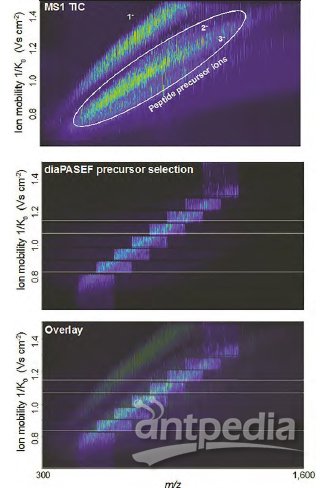
图4:该示例展示了dia-PASEF如何有效碎裂给定保留时间内释放出的所有肽离子。
数据提供:F. Meier, A.-D. Brumer, M. Frank, A.Ha, I.Bludau, E. Voytik, S. Kaspar-Schoenefeld, M. Lubeck, O. Raether, R.Aebersold, B. Collins, H.-L. Rost and M. Mann ,“Parallel accumulation – serialfragmentation combined with data-independent acquisition (diaPASEF): Bottom-upproteomics with near optimal usage”,bioRxiv 656207(2020)。https://doi.org/10.1101/656207
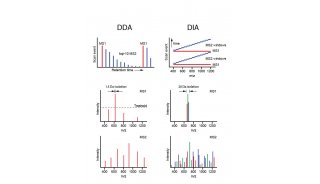
数据非依赖性采集
数据非依赖性采集(DIA)灵敏度的提升,激励MPIB计算系统生物化学团队使用机器学习算法将DIA工作流程集成到MaxQuant中。DIA的成功依赖于关键的仪器功能,即保证采集速度前提下的分辨率、灵敏度、准确度和动态范围。
研究人员使用timsTOF Pro进行实验时,通过利用TIMS和PASEF的速度和灵敏度来实现DIA测量,这一全新的DIA方法被称为dia-PASEF®。dia-PASEF®所生成的4D数据提供的离子淌度信息,帮助软件开发人员进行特征的对齐和提取。这些最新的技术进步以及DIA方法的发展为MPIB计算系统生物化学研究小组提供了新机遇。
由于仪器灵敏度的提高,DDA和DIA变得具有可比性。因此MaxQuant开发人员非常看好即将推出的DIA性能。
硬件操作对用户来说已经越来越简化,但是数据分析却更具挑战性,因为软件必须分辨出哪些碎片属于哪个分子。如前所述,解决这些挑战将提供对蛋白质组学的更深入研究,扩展4D-蛋白质组学应用于临床研究的可行性。
结论
生物信息学软件开发是一个不断变化的领域,在未来会有更多的技术创新。随着分析仪器的发展,MPIB计算系统生物化学团队必须在软件方面处理更多的信息,因为这些仪器在不断扩展其动态范围和性能。
通过MPIB计算系统生物化学研究团队与其他机构和行业领导者之间的重要合作,可以对MaxQuant软件平台进行改进。这些合作包括旨在优化提高蛋白质组学技术的项目,以及MaxQuant提供了分析新兴硬件平台上获取的数据所需的计算工具。这些合作的结果将用于开发软件的未来版本,以优化4D-蛋白质组学工作流程。
参考文献
1. J. Coxand M. Mann, “Quantitative, high-resolution proteomics for data driven systems biology”, Ann.Rev. Biochem. 80, 273–299 (2011). https://doi.org/10.1146/annurevbiochem-061308-0932162.
2. J. Cox, N. Neuhauser, A.Michalski, R.A. Scheltema, J.V. Olsen and M.Mann, “Andromeda: a peptide search engine integrated into the MaxQuant environment”, J. Proteome Res.10(4), 1794–1805 (2011). https://doi.org/10.1021/pr101065j
3. F. Meier, A.D. Brunner, S.Koch, H.Koch, M. Lubeck, M. Krause, N.Goedecke, J. Decker, T. Kosinski, M.A.Park, N. Bache, O. Hoerning, J. Cox, O. Räther and M. Mann, “Online Parallel Accumulation-Serial Fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer”, Mol. Cell. Proteomics 17(12),2534–2545 (2018). https://doi.org/10.1074/mcp.TIR118.0009004.
4. V. Marx, “A dream of single-cell proteomics”, Nat. Methods 16, 809–812(2019). https://doi.org/10.1038/s41592-019-0540-6
作者:Jürgen Cox,德国马克斯·普朗克生物化学研究所,Gary Kruppa,布鲁克·道尔顿
本文发表于《Spectroscopy Europe》 ,Vol. 32 No. 6 Dec/Jan 2020,第14-16页



 400-6699-117 转 2005
400-6699-117 转 2005