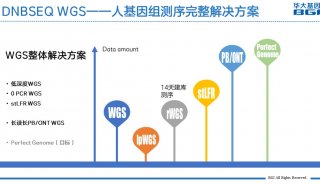
全基因组测序各步骤工具
全基因组测序(WGS)是下一代测序技术,用于快速,低成本地确定生物体的完整基因组序列。基因组的深度测序对于临床研究的意义重大,解读WGS数据并了解基因组突变在健康和疾病中的重要性是精准医疗的基石。
WGS分析流程能分为三大块,数据处理、检测变异和综合分析,具体如下图所示:
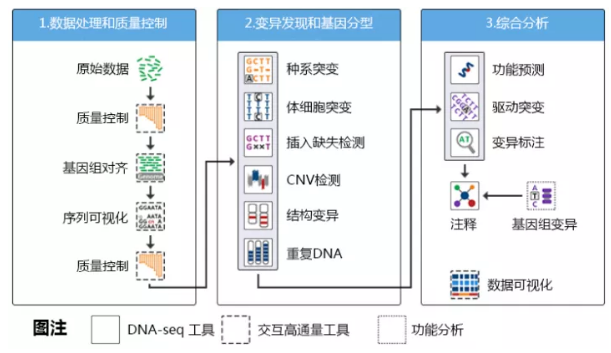
由于WGS现在已经非常成熟了,因此这里面的每一步骤都能在网上找到相应的工具。
质量控制
在高通量测序中,有时候会出现低质量读数和污染读数等问题,会影响接下去的分析结果。因此,质量控制(QC)对于原始NGS数据至关重要。现在质量控制软件主要用的是 NGS-QC Generator,它可以从与特定NGS概况相关联排序读数的分布推断出质量指标。
基因组对齐
高通量测序分析中需要将生成的读数对准(映射)到参考序列。此时,就需要用到Bowtie和BWA。前者能以每小时超过2500万个35 bp读数的速率将短DNA序列(读取)与人类基因组进行比对。后者针对大型参考基因组(如人类基因组)绘制低分歧序列。
序列可视化
可视化读取对齐是使用现有数据验证候选结构变体(SV)的最有效方式。这可以用IGV来完成。高性能的查看器可以有效地处理大型异构数据集,同时在所有基因组分辨率级别提供直观的用户体验。IGV的一个关键特征是其关注于基因组研究的综合性质,支持基于阵列和下一代测序数据,以及临床和表型数据的整合。

变异检测
都是各种基因变异的检测手段,就放一起说了。种系突变、体细胞突变和插入缺失检测这三个的检测都可以用一个软件,SAMtools。这是用于与高通量排序数据进行交互的程序。它可以处理SAM / BAM / CRAM格式的读取,写入,编辑,索引,查看和转换SAM / BAM / CRAM格式。SAMtools还可以索引FASTA格式的引用序列或从索引引用序列中提取子序列。

拷贝数变异(CNV)是遗传变异的常见来源,涉及许多基因组障碍。CNV是基因组中结构变异(SV)的一种形式。通常,CNV是指大于1kbp的DNA片段的重复或缺失。CNV检测可以用CNVnator,它可以从家族和群体基因组测序中发现基因型、表征典型和非典型CNV的方法。CNVnator具有灵敏度高(86%-96%),假阳性低(3%-20%),基因分型准确率高(93%-95%)的特点。
短串联重复序列(STR)是DNA重复的一种,重复单位为2-6bp,重复次数10~60多次,基因片段,400bp以下。STR是存在于人类基因组DNA中的一类具有长度多态性的DNA序列,不同数目的核心序列呈串联重复排列,而呈现出长度多态性,通常多态性片段长度在100-300bp。
STR的长度变异性与许多物种的表型变异有关,一些疾病也是由重复扩张引起。分析STR,特别是长STRs的变化是理解其个体变异性和导至其不稳定的机制的重要步骤。
检测STR比较简单,不用下软件,直接在线操作。用MISA-web (http://webblast.ipk-gatersleben.de/misa/) 中的MIcroSAtellite identification tool可以通过在输入字段中指定相应的登录号来从NCBI数据库中检索序列。MISA-web支持两种不同的输出格式:专有的MISA输出格式和通用GFF3。GFF3输出格式有助于将MISA网页搜索结果集成到下游分析中。
功能预测
现代测序技术产生越来越详细的基因组变异数据。然而,鉴于许多疾病性状复杂,是多基因联合作用,将单个变体或突变基因与表型相关联的常规方法已有其局限性。做功能基因预测的工具就比较多了。
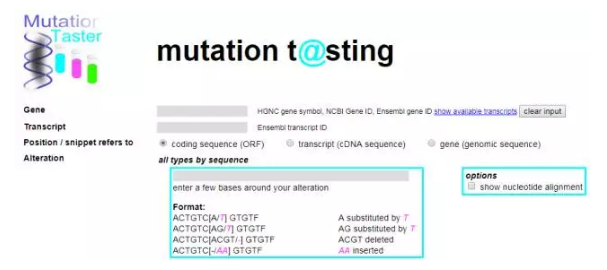
第一个在线工具是MutationTaster (http://www.mutationtaster.org/)。这个免费的在线应用可以快速评估DNA序列改变的致病潜力。
MutationTaster集成了来自不同生物医学数据库的信息,并使用已建立的分析工具。分析包括进化保守,剪接位点变化,蛋白质特征的丧失和可能影响mRNA量的变化。然后通过一个贝叶斯分类器评估测试结果,该分析器预测疾病潜力,一般的查询在0.3秒内就可完成。
第二个网站是SNPdryad(http://snps.ccbr.utoronto.ca:8080/SNPdryad/ ),可预测在人类蛋白质中氨基酸取代会发生的有害作用。而且在准确预测有害nsSNP方面,SNPdryad优于其他算法。

第三个是个数据集,dbNSFP(https://sites.google.com/site/jpopgen/dbNSFP),为人类非同义单核苷酸变体(nsSNV)和剪接位点变体(ssSNV)的功能预测和注释提供一站式资源,还有从外显子中发现的大量SNV筛选和测序研究。dbNSFP创建了基于人类参考序列的所有潜在nsSNV和ssSNV的列表,并为每个SNV编译了功能预测和注释。
目前该数据集中包括82,832,027 个nsSNV和ssSNV,附加的数据库dbscSNV编译所有潜在人类SNV及其有害性预测,另外增加了的15,030,459个潜在功能SNV。
驱动突变
区分驱动突变与偶发突变对于了解致癌分子机制,以及鉴定预后和寻找治疗靶标至关重要。MutSig可以分析在DNA测序中发现的突变列表,以便识别基因突变是驱动突变还是偶发突变。该软件最初是用于分析体细胞突变,但也可用于分析种系突变。MutSig建立了在肿瘤形成期间突变过程的模型,分析每个基因,以鉴定比预期突变更频繁的基因。
变异注释
最新的测序仪器与SNP发现工具结合可用于识别大量可能的SNP,而在识别后的下一个问题就是注释和选择功能重要的SNP。这项任务也能用dbNSFP完成,不过更推荐用ANNOVAR。它可以利用不断更新的信息来注释不同基因组(包括人类基因组hg18,hg19,hg38以及小鼠,蠕虫,蝇,酵母和许多其他基因组)检测到的遗传变异相关功能。ANNOVAR可以在一天内处理数百种人类基因组。
另外还有一个在线工具GeneTalk(http://www.gene-talk.de/),GeneTalk为分析人类序列变异的遗传学家提供了直观的基于网络的界面。它帮助临床遗传学家,寻找有关特定序列变异的信息,并将该用户与研究相同序列变异的其他用户连接起来。

数据可视化
基因组浏览器不仅用于显示最终结果,还能用于改进分析方法,测试数据质量和生成结果草图。有助于创建和利用基因组浏览器新进展来改进分析结果并支持基因组数据的快速可视化。
ZENBU这个数据集,通过数据处理和交互式链接实现可视化数据挖掘,使用户可以用BAM或制表符分隔(BED,GFF)序列对齐数据。

当然,除了这个在线工具外,前面介绍的IGV也能完成这个工作。
至此,一个完整WGS测序也就完成了。在临床研究中,目前火热的“精准医疗”和“个体化医学”都需要准确的基因组参考序列,来帮助真正“精准”的基因组数据分析。WGS测序不论是在科研还是临床诊疗上,今后都大有作为。