
欧洲制剂注册
为制剂药品欧洲注册申报及后续维护提供专业的、一站式、全生命周期的注册、GMP 咨询服务。
(1)在项目立项过程中,调研、评估制剂药品欧洲注册的可行性
(2)在项目研发过程中,提供专业的技术和法规支持
(3)欧洲GMP符合检查与指导、协助客户完成整改
(4)申报资料的翻译和撰写,保证申报资料符合欧洲注册要求
······
一、 服务范围
为制剂药品欧洲注册申报及后续维护提供专业的、一站式、全生命周期的注册、GMP 咨询服务。
二、服务内容
申报前
(1)在项目立项过程中,调研、评估制剂药品欧洲注册的可行性
(2)在项目研发过程中,提供专业的技术和法规支持
(3)欧洲GMP符合检查与指导、协助客户完成整改
(4)申报资料的翻译和撰写,保证申报资料符合欧洲注册要求
(5)eCTD文件的制作
申报
(1)与欧洲审评机构沟通、协助开展会议申请、预约递交
(2)递交申报资料、跟踪审评进度、指导发补回复
(3)为欧洲GMP现场检查提供翻译以及技术支持
(4)如需要,可提供欧洲境内的上市许可持有人、进口商、QP、QPPV服务
申报后
(1)产品获批后的变更维护服务(Type IA、IB、II)
(2)产品获批后PV系统维护服务
(3)产品获批后的再注册服务
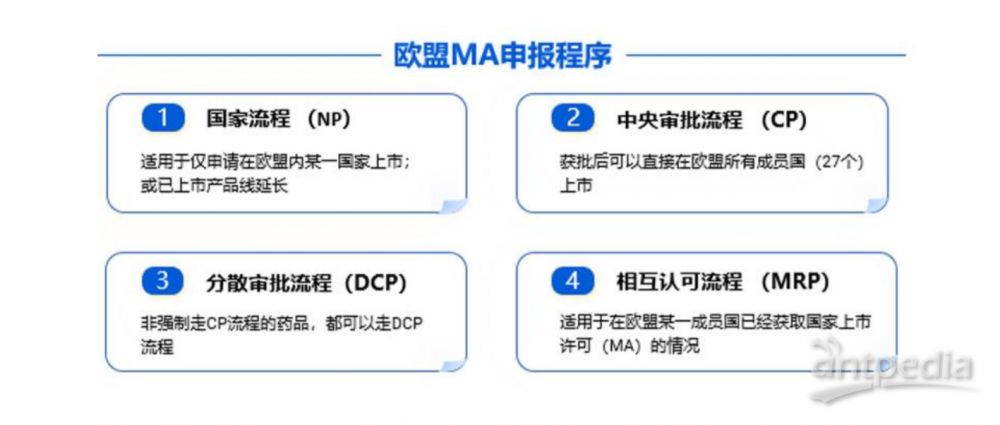
三、制剂申报途径

四、注册审评流程(以 DCP 为例)

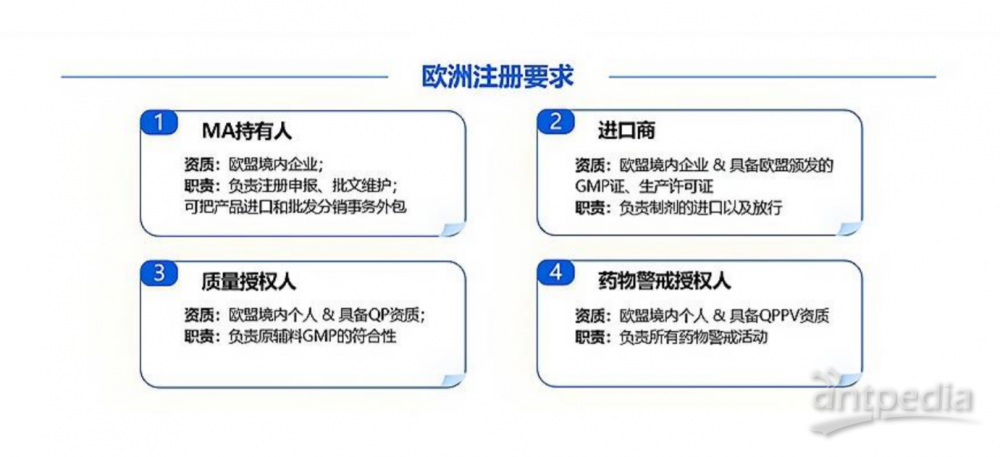
五、欧洲注册特别要求
欧盟 CEP/COS 申请
为符合欧洲药典各论或通则的原料药或辅料(通过合成、提取或发酵获得的有机或无机物)、具有 TSE 风险的产品、草 药及草药制剂提供在欧盟的 CEP/COS 首次申报、缺陷答复、变更递交、CEP 证书再注册等服务。
(1)提供CEP的基础培训,包括CEP申请介绍、CEP编写大纲等;
(2)提供中文CEP的编写大纲,负责对中文CEP进行差距分析、风险评估和法规指导;
(3)负责编写英文CEP,制作eCTD格式文件
一、服务范围
为符合欧洲药典各论或通则的原料药或辅料(通过合成、提取或发酵获得的有机或无机物)、具有 TSE 风险的产品、草 药及草药制剂提供在欧盟的 CEP/COS 首次申报、缺陷答复、变更递交、CEP 证书再注册等服务。
二、服务内容
CEP首次申报
(1)提供CEP的基础培训,包括CEP申请介绍、CEP编写大纲等;
(2)提供中文CEP的编写大纲,负责对中文CEP进行差距分析、风险评估和法规指导;
(3)负责编写英文CEP,制作eCTD格式文件;
(4)负责通过CESP通道递交CEP至EDQM;
(5)负责与EDQM的技术联络,并帮助客户关闭CEP缺陷以获得CEP证书;
(6)提供已获批的CEP申请姊妹CEP(CEP Sister File)服务;
(7)如有需要,可协助GMP部门为现场核查提供服务。
CEP日常维护
(1)提供已获批的CEP变更编写及递交、变更缺陷回复服务,直至变更获批;
(2)提供已获批的CEP证书再注册申请(Renewal)及递交服务。
三、欧盟 CEP/COS 简介
欧洲药典适用性证书(Certificate of Suitability to Monograph of European Pharmacopoeia),以前简称 COS,现在简 称 CEP。是由 EDQM 颁发的用以证明原料药的质量是按照欧洲药典各论描述的方法严格控制的,其产品质量符合欧洲 药典标准的一种证书。
经 EDQM 审批通过的原料药,将获得 CEP 证书。CEP 证书不仅得到所有欧盟成员国的认可,也得到了其他国家的承认, 例如加拿大、澳大利亚、新西兰、突尼斯和摩洛哥等。
CEP 证书可以分为以下几类:

四、CEP 首次申报注册流程

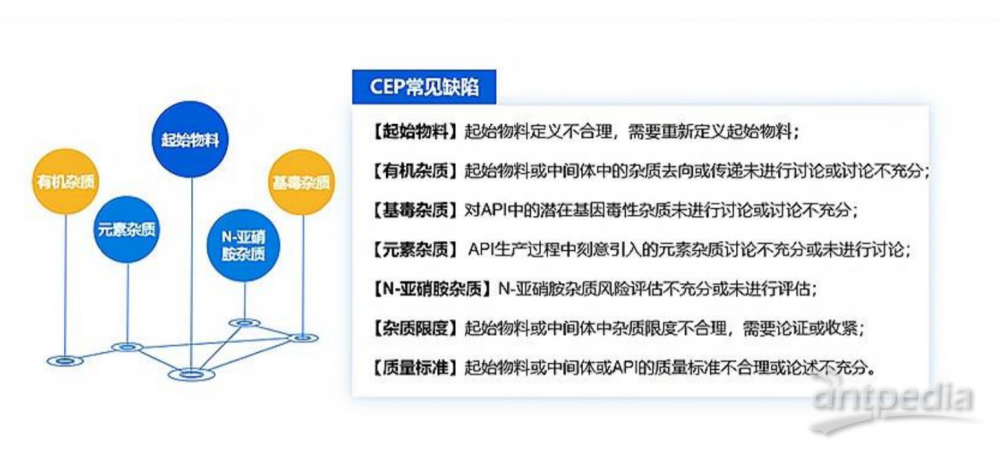
五、CEP 常见缺陷
欧盟 ASMF/EDMF 文件制作
为新的或现有的原料药(包含或未包含在 EP 药典或 EU 成员国药典中均可)提供在欧盟的 ASMF/EDMF 首次申报、缺 陷答复、变更递交等服务。
(1)提供ASMF的基础培训,包括ASMF申请介绍、ASMF编写大纲培训等;
(2)提供中文ASMF的编写大纲,负责对中文ASMF的差距分析、风险评估和法规指导;
(3)负责编写英文ASMF,制作eCTD格式文件;
(4)提供建立CESP递交通道的服务;
一、服务范围
为新的或现有的原料药(包含或未包含在 EP 药典或 EU 成员国药典中均可)提供在欧盟的 ASMF/EDMF 首次申报、缺 陷答复、变更递交等服务。
二、服务内容
(1)提供ASMF的基础培训,包括ASMF申请介绍、ASMF编写大纲培训等;
(2)提供中文ASMF的编写大纲,负责对中文ASMF的差距分析、风险评估和法规指导;
(3)负责编写英文ASMF,制作eCTD格式文件;
(4)提供建立CESP递交通道的服务;
(5)负责通过CESP通道递交至各国官方;
(6)负责与官方的技术联络,并帮助客户关闭ASMF缺陷直至完善;
(7)提供已完善的ASMF变更梳理、编写及递交;
(8)如有需要,可协助GMP部门为现场核查提供服务。
三、欧盟 ASMF/EDMF 简介
活性物质主文件(ASMF,Active Substance Master File),以前称为欧洲药物主文件(EDMF,European Drug Master File), 是由原料药生产商向 EMA 或欧盟各成员国药政递交的包含原料药保密信息的技术文件,用于支持制剂生产商的上市许 可申请。ASMF 的申请必须与使用该原料药的制剂的上市许可申请同时进行,其内容可分为公开部分和保密部分。
ASMF 的审评流程与索引该 ASMF 的制剂上市许可申报程序一致,具体分为以下几种:
四、欧盟 ASMF 的注册流程

五、ASMF 的常见缺陷

美国 IND 申请
提供试验性新药(Investigational New Drug,IND)在美国的 Pre-IND 会议申请、IND 申请、IND 维护(临床方案增补、 信息增补、安全性报告、年报报告)以及孤儿药认定(Orphan Drug Designations,ODD)申请等服务。
(1)提供Pre-IND会议申请的资料清单;
(2)负责对中文资料的差距分析、风险评估和法规指导;
(3)负责编写和制作英文Pre-IND meeting request和meeting package;
······
一、服务范围
提供试验性新药(Investigational New Drug,IND)在美国的 Pre-IND 会议申请、IND 申请、IND 维护(临床方案增补、 信息增补、安全性报告、年报报告)以及孤儿药认定(Orphan Drug Designations,ODD)申请等服务。
二、服务内容
Pre-IND申请
(1)提供Pre-IND会议申请的资料清单;
(2)负责对中文资料的差距分析、风险评估和法规指导;
(3)负责编写和制作英文Pre-IND meeting request和meeting package;
(4)提供美国本土代理及ESG通道服务,以递交Pre-IND资料至FDA;
(5)负责与FDA的技术联络,并帮助客户顺利拿到Pre-IND官方咨询答复。
IND申请
(1)提供IND申请的基础培训和资料清单;
(2)负责对CMC、非临床和临床资料的差距分析、风险评估和法规指导;
(3)负责按CTD各模块编写和制作IND的英文申请资料,并制作eCTD;
(4)提供美国本土代理及ESG通道服务,以递交IND资料至FDA;
(5)负责与FDA的技术联络,并帮助客户顺利通过IND申请;
(6)提供IND维护(临床方案增补、信息增补、安全性报告、年报报告)服务;
(7)如有需要,可协助GMP部门为现场核查提供服务。
ODD认定
(1)负责对拟申请产品的相关调研,评估该产品申请孤儿药认定的可行性;
(2)提供资料清单并对中文资料进行差距分析、风险评估和法规指导;
(3)负责编写和制作孤儿药认定的英文申请资料;
(4)提供美国本土代理服务,以递交孤儿药认定资料至FDA;
(5)负责与FDA的技术联络,并帮助客户顺利拿到孤儿药资格。
三、美国 IND 简介
试验性新药(Investigational New Drug,IND):一般指尚未经过上市批准、正在进行各阶段临床研究的新药。CFR (美国 联邦法规) 禁止任何未经 FDA 批准的新药在州际间运输交易,因此 IND 申请的目的是获得未批准药物用于人类临床试 验的许可,而在法律上讲是申请 IND 药物的调运许可。 IND 申请之前可申请 Pre-IND 会议,就不同学科的问题寻求 FDA 的专业意见;IND 申请递交后经 30 个日历日审评,如 FDA 无意见,则 IND 默认生效;美国一个 IND 号贯穿整个 IND 生命周期,可在不同阶段通过增补或年报等形式对 IND 进行维护;与此同时,美国《孤儿药法案》(Orphan Drug Act,ODA)鼓励企业研究和开发孤儿药,在 IND 申请前后 申请孤儿药资格,可争取到更多的政策福利。

四、美国 pre-IND 会议申请流程
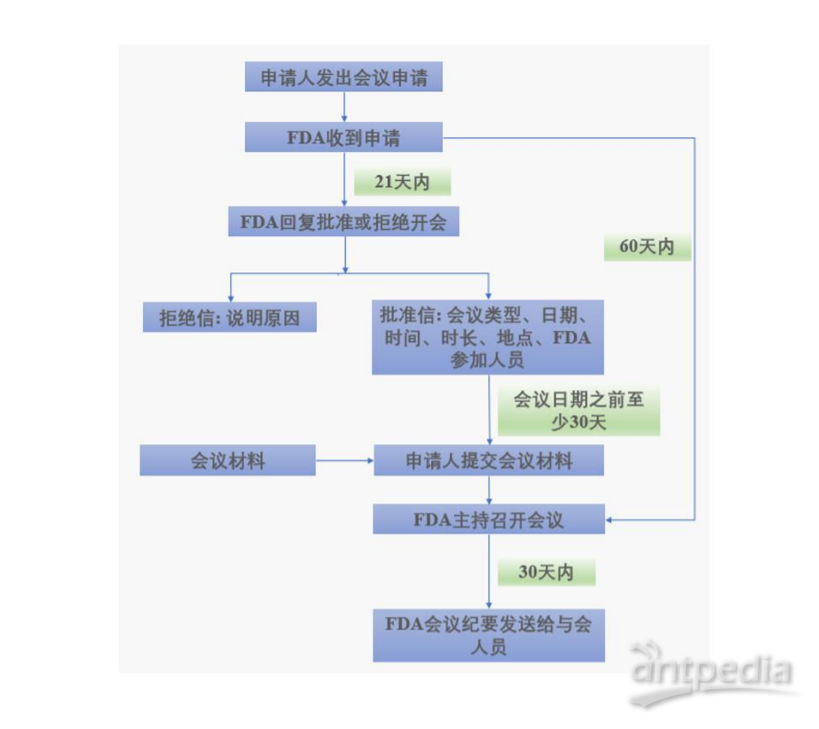
五、美国 IND 申请流程

六、美国孤儿药认定申请流程

美国NDA
为美国新药申请(New Drug Application,NDA)及后续维护提供专业的、全生命周期的注册、GMP 咨询服务。
(1)提供美国注册代理服务;
(2)提供Pre-NDA会议申请、会议资料准备及递交等服务;
(3)负责对各学科资料(CMC、非临床和临床)的差距分析、风险评估和法规指导;
(4)负责按CTD各模块编写、翻译和制作英文NDA资料,并制作eCTD;
一、服务范围
为美国新药申请(New Drug Application,NDA)及后续维护提供专业的、全生命周期的注册、GMP 咨询服务。
二、 服务内容
(1)提供美国注册代理服务;
(2)提供Pre-NDA会议申请、会议资料准备及递交等服务;
(3)负责对各学科资料(CMC、非临床和临床)的差距分析、风险评估和法规指导;
(4)负责按CTD各模块编写、翻译和制作英文NDA资料,并制作eCTD;
(5)提供ESG递交通道服务,负责将NDA递交至FDA;
(6)负责跟踪FDA审评进度,积极与FDA审评PM沟通联络;
(7)负责指导客户关闭FDA在审评过程中提出的各类缺陷(IRL、DRL、CRL等);
(8)提供NDA的日常维护服务,包括变更/增补、年报等;
(9)提供为生产厂址申请DUNS号、FEI号的服务;
(10)如有需要,可协助GMP部门为现场核查提供服务。
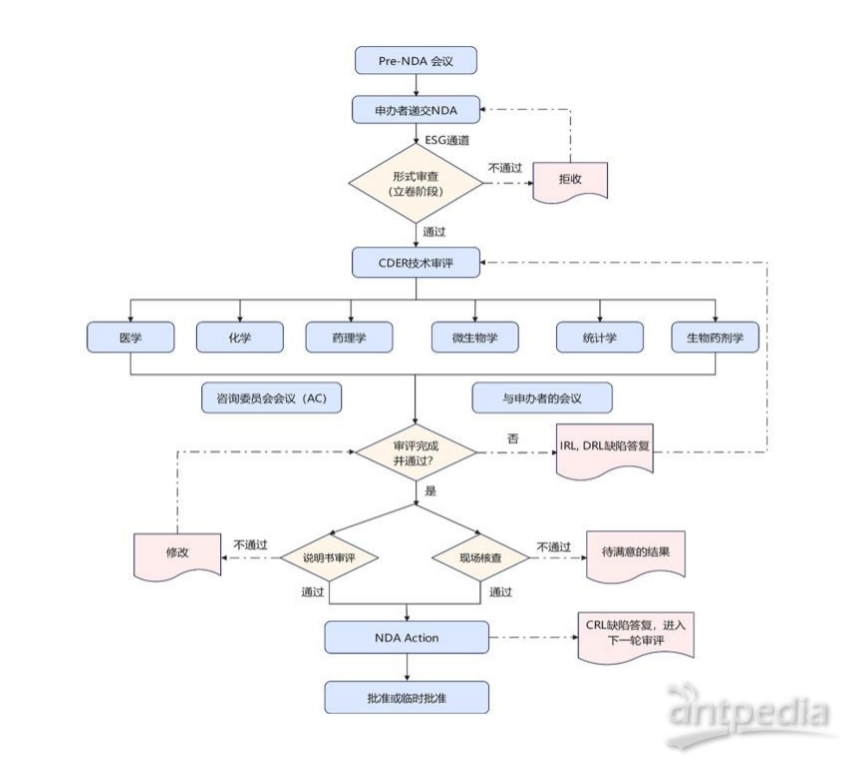
三、美国 NDA 简介
当申办者有足够数据(包括 CMC、非临床和临床数据)能证明药品的安全性和有效性满足了 FDA 对于上市的要求时, 申办者就可以向 FDA 递交新药申请(New Drug Application,NDA)。依据《食品、药品和化妆品法》(FD&C Act), 一般符合以下情况均可向 FDA 提出 NDA 申请:

依据处 方药申报者付费法案(Prescription Drug User Fee Act, PDUFA),NDA 是需要缴费的,其中 2021-2022 财年的 费用如下:

四、美国 NDA 注册流程
美国 ANDA
提供仿制药在美国的 ANDA 首次申报、缺陷答复(直至获批)、ANDA 变更及年报维护等服务。
(1)在前期项目立项过程中,调研、评估分析ANDA注册的可行性
(2)在项目研发过程中,提供专业的技术、法规和现场支持
(3)美国cGMP符合性检查与指导、协助客户完成整改
(4)申报资料的翻译和撰写(除BE外),保证申报资料符合ANDA注册要求
......
美国 DMF 文件制作
提供原料药、原料药中间体、包装材料、辅料等产品在美国的 DMF 首次申报、缺陷答复(直至通过 FDA 审评)、DMF 增补(行政和/或质量变更)、年度报告等服务。
(1)提供DMF的基础培训,包括美国DMF的介绍、管理制度等;
(2)提供中文DMF编写大纲,负责对中文DMF进行差距分析、风险评估和法规指导;
(3)负责编写英文DMF,制作eCTD格式文件,及ESG递交申报资料至FDA;
......
FDA 工厂注册、自认定与 NDC 申请
FDA 工厂注册简介
FDA 工厂注册,即“Establishment Registration”,又叫场地注册。产品登记,即“Listing”。
根据美国联邦食品药品化妆品管理法和美国联邦法规相关要求,任何从事药品生产和包装活动的企业必须将其生产地址 与产品在 FDA 登记。此项要求对美国本土企业和海外企业同样适用。 对于海外企业,工厂注册和产品登记是美国 FDA 对进口药品的重要监管措施之一。FDA 可以通过注册号第一时间查到 相关企业的详细信息,并可及时联系相关企业在美国的代理人,有效掌握相关企业产品在美信息。
......

 2850412321
2850412321













