-
Xalibur软件常见的问题及解决方法
红杏 发布于 2008-02-11 02:17:31
转帖
1、通讯问题
1.1 网线(直通线,前期买的都为黄色的线)或switch (转接器,接网线的)连接松了。
解决方案:拔掉重接
1.2 IP地址的更改
所有仪器的IP地址均为:172.16.0.101,子网地址:255.255.0.0
1.3 网卡被禁用(Disable)
2、系列运行不正常
2.1 有错误提示的,先按Printscreen拷屏,如有错误代码的,记下错误代码。
2.2 看各个模块(泵,自动进样器及质谱)的工作状态(Busy,intializing,downloading,or wainting for contact)
2.3 系列应重新建,在原来的基础上建容易出问题2.4 重装软件
2.4.1 ghost, 备份重要的方法和数据2.4.2 重装软件:
a、备份重要的方法和数据到其他逻辑盘
b、最新的校正方法(Caibration file)应备份
c、从控制面版依次删除:TSQ Quantum, Xcalibur LC Devices, Xcalibur等软件)
d、重启,重新安装
3、文件打不开
3.1 Xcalibur高版本采集的数据低版本打不开
3.2 最近采集的数据如文件大小太少打不开,可能没采集上数据,可重新采集
4、 常见的错误信息
4.1 机械故障:找不到瓶子,传感器坏了等
4.2 有错误代码的,拷屏并记下错误代码。 -
Orbitrap:质谱技术的重大突破,赛默飞世尔再创辉煌
阿土仔 发布于 2008-09-09 20:34:03
——访赛默飞世尔科技的Orbitrap质谱仪发明人马可洛夫博士
人类文明的发展越来越体现在对物质世界认识深化的科学和技术的进步上。质谱仪作为人类深入了解客观世界的主要工具之一,日益受到重视。质谱技术是分析技术中发展最快的。10年前中国质谱仪的拥有量仅在一百台左右,而今天已超过千台,用户对质谱仪的要求也在不断地提高。各大分析仪器公司无不投巨资激励科研人员创新技术提高质谱仪的性能,以满足客户的需求, 如飞行时间,离子阱技术都得到了一定的发展。在现有质谱技术中,傅里叶变换离子回旋共振质谱FT-MS的性能无疑是最强的, 然而其价格和运行维护成本也出现了直线的攀升,使客户欲拥有之又望尔却之。鱼和熊掌不能兼得,这似乎成了质谱仪发展的一条规律。然而,赛默飞世尔科技推出的静电场轨道阱(Orbitrap)质谱仪改写了这一规律使之成为历史。
Orbitrap实现了二十年来质谱仪发展的革命性突破,在争攀高峰的质谱仪赛场上,犹如一匹腾飞的黑马越过所有的竞争对手,登上了一个全新的高峰。它的性能可与傅里叶变换离子回旋共振质谱FT-MS媲美,而它的价格尤其是运行维护成本则几乎达到了普及型仪器的水平,所实现的性/价比是其他同类仪器所望尘莫及的,这使它成为每个客户都值得拥有的鱼和熊掌兼得的白马王子。赛默飞世尔推出的新款复合型线性离子阱静电场轨道阱质谱仪(LTQ Orbitrap)一举夺得了2006年的Pittcon金奖(Gold award at Pittcon 2006)和2006年的百名世界研发精英奖(R&D 100 award for 2006)。Orbitrap质谱仪的发明人亚力山大 马可洛夫博士也因此在2007年获得了默克奖(Heinrich Emanuel Award)和俄罗斯质谱学会的金牌(Gold Medal from Russian for Mass Spectrometry)。
《生物技术世界》杂志有幸专访了马可洛夫博士。通过专访我们了解了发明人的创新历程,赛默飞世尔科技对他进行大力支持所取得的成就,以及赛默飞世尔科技向客户提供的优质服务和勇于开拓应用的强者气魄。借此报道的机会我们将此次专访中获得的,有关这位带传奇色彩的发明天才科学家的创新历程和赛默飞世尔科技的成就的主要内容整理出来与读者分享。
发明天才的创新历程
亚力山大 马可洛夫(Alexander Makarov)1966年出生在俄罗斯Irkutsk的Siberian镇。从童年时代起他就对技术感兴趣,总是试图发明各种各样的技术。然而,那些发明不是被证明为不现实就是早已有人作了同样的发明。不过马可洛夫的天才爱好将他带入了莫斯科工程物理学院,一个符合他的理想去追求应用而不是基础科学的地方。
在大学时代,马可洛夫对飞行时间质谱仪产生了兴趣。他的硕士和博士论文都是研究飞行时间质谱的。1992年他在莫斯科工程物理学院通过博士论文答辩之后,自己安排了一次访问几所英国大学的旅行讲学,在他博士论文的基础上给学生上课。在这次旅行中,他引起了英国曼切斯特的Kratos Analytical公司的注意,结果他成了Kratos的离子光学顾问。但由于Kratos的雇佣冻结,他返回了莫斯科。由这段经历所建立起来的与英国方面的联系后来帮助他成了英国Warwick大学的一名博士后,在那里他开始了串联质谱的研究。
1996年他加盟了英国曼切斯特的一家小型高技术公司HD Technologies,并在那里开始研究Orbitrap质谱。他从英国政府赢得了SMART奖金,用于购买实验器材。在同事们的帮助下,马可洛夫自己动手,建立了一个使用激光源的实验设备。在1998年10月获得第一个质谱之后,这个实验设备显示了增加高分辨率的能力。1999年该设备实现了分辨率160,000。
2000年原热电公司购并了HD Technologies。这一事件使得马可洛夫博士获得了专门研究Orbitrap的机会。原热电公司为他组建了研究团队。一年之后获得了第一个成果,使用仅有的一个外部RF存储器来积累大量的离子并将它们快速地射入Orbitrap质量分析器。研究工作开始向着研发实际应用的质谱仪发展。随着研发的深入,2002年7月,原热电公司关闭了曼切斯特的工厂,将Orbitrap的研发工作转移到了Bremen,又为研发团队增加了项目经理,机械工程师,固件专家,电子工程师等各类人员。
最后,这项工作取得了令人惊喜的结果,在San Antonio的ASMS大会上推出了LTQ Orbitrap,实现了质谱仪发展的革命性突破。马可洛夫博士说:“Orbitrap是第一个在静电场中进行离子捕获的高性能质量分析器。LTQ Orbitrap是第一台实现Orbitrap分析的组合质谱,其中主要由线性离子阱实现离子分离、裂解和多级质谱MS_功能,而Orbitrap则对随后的离子包提供精确的质量分析和高分辨率检测,这之间通过一个反射曲线多极的C形离子阱(C-trap)相连。”
金奖质谱仪所洋溢的威力
赛默飞世尔科技推出的两款新的LTQ Orbitrap,LTQ Orbitrap Discovery和LTQ Orbitrap XL,提供了出色的质量精确度、分辨能力、动态范围和灵敏度。在复杂蛋白鉴定能力和灵敏度方面,提供了超越四级杆-飞行时间质谱(Q-TOF)的巨大优势。它们是制药业、代谢组学、蛋白组学和生物标记发现等研究领域的理想适用仪器。
LTQ Orbitrap Discovery是高度灵敏的系统,具有高达30K的分辨能力和一个超常的扫描率,具有轨迹层分离的能力。所提供的高分辨率和精确质量鉴定能力使它能够对差别表达完成高通量的无标志序列分析,而它的MSn能力又使它成为小分子特性鉴定和结构阐明的理想仪器。
LTQ Orbitrap XL是蛋白鉴定和生物标记发现的优秀平台,具有无可超越的MS和MSn的灵敏度、快速扫描率、高质量精确度和高达100K的分辨能力,并具有新的HCD八极碰撞单元的特性,可以在MS/MS裂解应用中增加灵活性,包括基于iTRAQ的多肽定量,翻译后修饰(PTM)分析,从头测序和代谢组学研究等应用。作为蛋白组学平台也可以兼容或通过升级来使用电子转移裂解(ETD)选项,用于肽和蛋白的有控制的裂解。它对先进的蛋白组研究来说是不可或缺的。
马可洛夫博士说:“Orbitrap提供的高分辨率和精确质量鉴定能力加上仪器前台的LTQ线性离子阱有格外高的速度、灵敏度和MSn能力,使得 LTQ Orbitrap质谱仪的性能如此优异,独占鳌头。”
使新药发现更有希望
大量的临床前期研究要求对代谢谱进行详细分析,以进行新药的质量认证,因而具备在线精确质量测定能力的LC/MS及MSn分析是代谢物鉴定的重要前提。LTQ Orbitrap可以产生多极串联质谱数据,具备超fmol(10-15 mol)级灵敏度、高质量精确度以及其他多种离子化模式。还可整合使用一个完整的样品制备协议和一个新的软件工具SIEVE,该软件能够对大型LC/MS数据集进行差别比较分析,使得代谢物、蛋白和多肽的无标志、半定量差别表达自动化。如果再将此和Mass Frontier软件相结合,也就是将鉴定代谢物差别表达的SIEVE软件包和鉴定代谢物结构的Mass Frontier软件包无缝整合,就可以高可信度鉴定代谢物及其结构。
在蛋白质组学研究中,分析复杂的蛋白质酶解物是质谱分析面临的最艰巨任务之一,高分辨的精确质量数测定和宽动态范围是进行可靠蛋白鉴定的重要技术前提。如血液样品中有丰富的白蛋白,同时又含有浓度低数万亿倍的其他重要生物标记物,通过分级分离可以简化低丰度生物标记物的检测、鉴定和分析,这个过程主要是将样品连续不断地引入质谱仪,将信号作均一化的处理,从而能够从中采集到足够的信息图谱并准确地测定质量数,最终获得生物标记物的结构信息。LTQ Orbitrap的质量测定和动态排除能力,以及优异的序列覆盖率可以保证可靠的低丰度蛋白质鉴定。
马可洛夫博士说:“LTQ Orbitrap 的突出性能使之在代谢组学研究和蛋白质组学中“自下而上”(Bottom-Up)包括最近发展的“从中向下”(Middle-Down)的研究方法中更具优势,极大地改善可靠性、分析速度,和分析物鉴定的检测限制。”
对2008奥运会的优质服务
尽管赛默飞世尔科技不断地在推出顶级产品,然而适用的是最好的。一直关注着2008北京奥运会的赛默飞世尔搜尽产品宝库,从兴奋剂检测、环境监测,到机场反恐安全和绿色奥运,一一捧出了最适用仪器和解决方案的优质服务。
在2008北京奥运会兴奋剂检测仪的公开招标中,赛默飞世尔从众多竞争对手中脱颖而出,为国家兴奋剂检测中心提供了一台DFS高分辨率质谱仪。该仪器的灵敏度超过同类产品的8~9倍,在要求一万以上分辨率的二恶英分析和兴奋剂分析中是灵敏度最高的质谱,可以同时实现药物检测的快速筛选和确认分析。同时,它的自动化程度高,使用简单方便,而能耗只有同类产品的几分之一。
对2008奥运会来说,北京的空气质量将成为保障奥运会顺利开展的关键。赛默飞世尔为中国环境科学研究院提供了二套空气质量监测系统,用于评估北京奥运期间大气污染控制措施的效果。针对北京空气质量的特点,赛默飞世尔为所提供的监测系统作了专门的配置,还安装了校准设备和数据采集装置,使其能够提供空气质量的实时监测,数据采集和数据传输,并能实现不同灵敏度的在线连续监测。
随着奥运反恐安保日益得到重视,北京、上海、成都、广州等多个机场已经陆续开始安装赛默飞世尔的SGS辐射探测系统,用来监测和阻止非法核武器和放射性材料通过机场进入中国。由于机场的所有检测都不得过度干扰合法的商业物流,安保人员需要在货物的行进中对其进行严密的检测,赛默飞世尔对SGS系统进行了优化,使其具备了快速清除误判和发现真实核威胁的能力,以满足现场执行的需要。
2008奥运会将向世界呈现一个绿色的奥运。这与赛默飞世尔科技致力于把世界变得更健康、更清洁、更安全的目标不期而遇。赛默飞世尔已经为此向国内食品安全监督机构提供了监控食品安全的技术支持。
创新革命夺金奖开拓应用价更高
在专访中,我们不仅看到了赛默飞世尔科技注重技术创新和为客户配置最适用仪器的优质服务的王者风范,而且还看到了赛默飞世尔勇于开拓应用向未知世界进军来实现更高价值的强者气魄。赛默飞世尔始终不渝地对已有的产品进行新的应用开拓,使之能够更好地为客户解决问题。马可洛夫博士说:“LTQ Orbitrap提供了一个功能非常强大的硬件平台,它所具备的能力和它正在被利用的能力之间相去甚远。将来的应用和软件开发旨在填补这一差距,从而使得这个硬件平台的通量,分析速度,额外的谱信息等能够更好的为分析所利用。”
由此我们是否可以想一下,现在有多少问题可以通过开发正确的应用和软件来发挥LTQ Orbitrap这个硬件平台目前还没有发挥出来的能力去加以解决呢?一定很多。例如:通过分析化合物的成份和结构或其代谢物的结构来发现新药,LTQ Orbitrap硬件平台和已有的软件已经能有上佳的表现。但是,当我们面向未知的复杂化合物,如复方的验方中药,LTQ Orbitrap硬件平台和已有的软件还难以直接通过分析验方的未知化合物和其代谢物来确定其中起药物作用的所有物质构成。面对这样的未知世界难题,是致力于创新威力更强大的质谱硬件吗?为此,人类也许要等上几十年、几百年。然而,赛默飞世尔科技已经为我们树立了榜样。赛默飞世尔在应用开发领域的投入和努力从未止步,他们最近与麻省通用医院(Massachusetts General Hospital)积极合作进行蛋白生物标记发现的研究。该医院的大量详细注释的血浆样本理想地适合于使用LTQ Orbitrap来进行生物标记发现的研究,突破在血浆样本中发现痕量生物标记的瓶颈,从而推进应用和软件开发的步伐。可见,开发正确的分析软件和数据库,更多更好地发挥LTQ Orbitrap硬件平台的能力,将来是完全有可能解读中药复方中起药物作用的物质构成的。《生物技术世界》衷心地希望赛默飞世尔科技及其广大的客户在开拓LTQ Orbitrap的应用和全面发挥这个硬件平台威力的努力中能够取得惊人的进展。
作者: 《生物技术世界》第2007-10期
本刊记者 孙建伟
-
绝对回收率和相对回收率
troy.tper.lee 发布于 2007-12-21 13:43:00
绝对回收率和相对回收率
这个问题我们实验室前一阵重点讨论过,我个人总结为:
(一)用于评价样品和内标等的回收程度,一般用绝对响应(峰面积或峰高)直结计算,与方法的灵敏度很相关。
绝对回收率:考察测定结果与“真值”的接近程度,从生物样本基质中回收得到目标物质的响应值除以纯标准品产生的响应值即为分析物的绝对回收率。应选用高、中、低三种浓度分别进行考察,绝对回收率一般要求在50%~80%(这个数值范围书上是这么写的,不过,个人认为接近100%更好,太低的话,比如说20%等,可能方法就不容易建立,此时,应更投提取体系,或者是采用反提法等)。
提取回收率:空白基质中定量加入药物,经处理后与空白基质处理后加入相同量药物的比值。绝对回收率与提取回收率的关系:
绝对回收率=提取回收率 / (1-基质效应)
对于液相色谱等而言,基质效应为0,此时,绝对回收率等于提取回收率。
(二)用于评价方法的准确度,等同于相对偏差的作用,用测定所得浓度结果去计算。
相对回收率(又称方法回收率),从生物样本基质中回收得到目标物质的响应值除以加入到生物基质中已知量的标准品产生的响应值即为分析物的相对回收率。该评价指标已扣除了目标药物在样品的前处理过程损失而造成的系统误差。高、中浓度点测得的相对回收率应在85~115%,低浓度(定量限)应在80~120%。回收率的具体操作
绝对回收率:空白基质中定量加入药物经处理后所测得峰面积 / 与基质相同体积水(血浆等我们一般用水代替)处理后加入相同量药物所测得峰面积*100%
提取回收率:空白基质中定量加入药物经处理后所测得峰面积 / 空白基质处理后加入相同量药物所测得峰面积*100%
基质效应:(1-空白基质处理后加入相同量药物所测得峰面积 / 与基质相同体积水(血浆等我们一般用水代替)处理后加入相同量药物所测得峰面积*100%)
上面三者知道其二,也可以用下面的公式计算剩余的:绝对回收率=提取回收率 / (1-基质效应)
相对回收率:测定浓度 / 标矢浓度 -
内标和外标是什么及其选用
firefox 发布于 2007-08-10 23:45:56
来源:仪器信息网
--------------------------------------------------------------------------------An internal standard should be used when performing MS quantitation. An appropriate internal standard will control for extraction, HPLC injection and ionization variability. In a complex matrix it is not uncommon for two different standard levels in SRM integrated plots, at the lower end of the standard curve, to give nearly an identical response. It is only when an internal standard is used that the two points can be differentiated. Some researchers attempt to prepare standard curves and run samples without an internal standard and find moderate success. Often without an internal standard % RSDs of replicates can be as high as 20%. Using an internal standard the % RSDs can be brought down to approximately 2%. We run triplicates at each level of our standard curve.
How do I choose an internal standard?
The best internal standard is an isotopically labeled version of the molecule you want to quantify. An isotopically labeled internal standard will have a similar extraction recovery, ionization response in ESI mass spectrometry, and a similar chromatographic retention time. If you are performing non-clinical PK quantitation it may be difficult to justify such a standard since a special synthesis of an isotopically labeled standard can be expensive and time consuming. Often if you are working with medicinal chemists they will have a library of compound analogs that can be used as internal standards. These analogs were made in the evolution of the compound to be tested and will be similar to the compound to be quantified and more importantly will be slightly different by parent mass. Try to avoid using de-methylated (-14) or hydroxylated (+16) analogs as internal standards since these are the most common mass shifts observed in naturally occuring metabolites of the parent compound. A common internal standard is a chlorinated version of the parent molecule. A chlorinated version of the parent molecule will commonly have a similar chromatographic retention time which is an important characteristic of an internal standard. We have found that one of the most important characteristics of an internal standard is that it co-elutes with the compound to be quantified.
How do I use an internal standard?
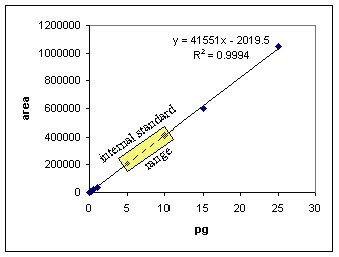
First of all an internal standard should be added at the beginning of the sample work-up, typically before the plasma crash or solid phase extraction. The internal standard should be added at the same level in every sample including the standards. An internal standard should give a reliable MS response. Care should be taken that the amount of the internal standard is well above the limit of quantitation but not so high as to suppress the ionization of the analyte. "How much internal standard should I add?", this is an important question. It pays to know roughly how much compound is in your sample. This can be accomplished by making trial analyses of an early, middle and late time point with perhaps one or two standard points. This information will be very valuable when building an appropriate standard curve and in knowing how much internal standard to add. If you were trying to quantify samples in the range of 100 fg to 25 pg and the limit of detection was 100 fg you might add 5 to 10 pg of internal standard to every sample. A good rule of thumb is to target the internal standard to the lower 1/3 of the working standard curve. This is a range that will give a comfortable response without interfering with the ionization of the analyte.
--------------------------------------------------------------------------------
中文:
什么叫内标法?怎样选择内标物?
内标法是一种间接或相对的校准方法。在分析测定样品中某组分含量时,加入一种内标物质以校谁和消除出于操作条件的波动而对分析结果产生的影响,以提高分析结果的准确度。
内标法在气相色谱定量分析中是一种重要的技术。使用内标法时,在样品中加入一定量的标准物质,它可被色谱拄所分离,又不受试样中其它组分峰的干扰,只要测定内标物和待测组分的峰面积与相对响应值,即可求出待测组分在样品中的百分含量。采用内标法定量时,内标物的选择是一项十分重要的工作。理想地说,内标物应当是一个能得到纯样的己知化合物,这样它能以准确、已知的量加到样品中去,它应当和被分析的样品组分有基本相同或尽可能一致的物理化学性质(如化学结构、极性、挥发度及在溶剂中的溶解度等)、色谱行为和响应特征,最好是被分析物质的一个同系物。当然,在色谱分析条什下,内标物必须能与样品中各组分充分分离。需要指出的是,在少数情况下,分析人员可能比较关心化台物在一个复杂过程中所得到的回收率,此时,他可以使用一种在这种过程中很容易被完全回收的化台物作内标,来测定感兴趣化合物的百分回收率,而不必遵循以上所说的选择原则。
在使用内标法定量时,有哪些因素会影响内标和被测组分的峰高或峰面积的比值?
影响内标和被测组分峰高或峰面积比值的因素主要有化学方面的、色谱方面的和仪器方面的三类。
由化学方面的原因产生的面积比的变化常常在分析重复样品时出现。
化学方面的因素包括:
1、内标物在样品里混合不好;
2、内标物和样品组分之间发生反应,
3、内标物纯度可变等。对于一个比较成熟的方法来说,色谱方面的问题发生的可能性更大一些,色谱上常见的一些问题(如渗漏)对绝对面积的影响比较大,对面积比的影响则要小一些,但如果绝对面积的变化已大到足以使面积比发生显著变化的程度,那么一定有某个重要的色谱问题存在,比如进样量改变太大,样品组分浓度和内标浓度之间有很大的差别,检测器非线性等。进样量应足够小并保持不变,这样才不致于造成检测器和积分装置饱和。如果认为方法比较可靠,而色谱固看来也是正常的话,应着重检查积分装置和设置、斜率和峰宽定位。对积分装置发生怀疑的最有力的证据是:面积比可变,而峰高比保持相对恒定,
在制作内标标准曲线时应注意什么?
在用内标法做色谱定量分析时,先配制一定重量比的被测组分和内标样品的混合物做色谱分析,测量峰面积,做重量比和面积比的关系曲线,此曲线即为标准曲线。在实际样品分析时所采用的色谱条件应尽可能与制作标准曲线时所用的条件一致,因此,在制作标准曲线时,不仅要注明色谱条件(如固定相、柱温、载气流速等),还应注明进样体积和内标物浓度。在制作内标标准曲线时,各点并不完全落在直线上,此时应求出面积比和重量比的比值与其平均位的标准偏差,在使用过程中应定期进行单点校正,若所得值与平均值的偏差小于2,曲线仍可使用,若大于2,则应重作曲线,如果曲线在铰短时期内即产生变动,则不宜使用内标法定量。
外标法
用待测组分的纯品作对照物质,以对照物质和样品中待测组分的响应信号相比较进行定量的方法称为外标法。此法可分为工作曲线法及外标一点法等。工作曲线法是用对照物质配制一系列浓度的对照品溶液确定工作曲线,求出斜率、截距。在完全相同的条件下,准确进样与对照品溶液相同体积的样品溶液,根据待测组分的信号,从标准曲线上查出其浓度,或用回归方程计算,工作曲线法也可以用外标二点法代替。通常截距应为零,若不等于零说明存在系统误差。工作曲线的截距为零时,可用外标一点法(直接比较法)定量。
外标一点法是用一种浓度的对照品溶液对比测定样品溶液中i组分的含量。将对照品溶液与样品溶液在相同条件下多次进样,测得峰面积的平均值,用下式计算样品中i组分的量:
W=A(W)/(A)
式中W与A分别代表在样品溶液进样体积中所含i组分的重量及相应的峰面积。(W)及(A)分别代表在对照品溶液进样体积中含纯品i组分的重量及相应峰面积。外标法方法简便,不需用校正因子,不论样品中其他组分是否出峰,均可对待测组分定量。但此法的准确性受进样重复性和实验条件稳定性的影响。此外,为了降低外标一点法的实验误差,应尽量使配制的对照品溶液的浓度与样品中组分的浓度相近。
外标法 external standard method 色谱分析中的一种定量方法,它不是把标准物质加入到被测样品中,而是在与被测样品相同的色谱条件下单独测定,把得到的色谱峰面积与被测组分的色谱峰面积进行比较求得被测组分的含量。外标物与被测组分同为一种物质但要求它有一定的纯度,分析时外标物的浓度应与被测物浓度相接近,以利于定量分析的准确性。
--------------------------------------------------------------------------------
定量分析中怎样选择内标法或外标法
选一与欲测组分相近但能完全分离的组分做内标物(当然是样品中没有的组分),然后配制欲测组分和内标物的混合标准溶液,进样得相对校正因子。再将内标物加入欲测组分的样品中,进样后测得欲测组分和内标物的定量参数。用内标法公式计算即可。
内标法是将一定量的纯物质作内标物,加入到准确称量的试样中,根据被测试样和内标物的质量比及其相应的色谱峰面积之比,来计算被测组分的含量。 选择内标物有4个要求:1.内标物应是该试样中不存在的纯物质;2.它必须完全溶于试样中,并与试样中各组分的色谱峰能完全分离;3.加入内标物的量应接近于被测组分;4.色谱峰的位置应与被测组分的色谱峰的位置相近,或在几个被测组分色谱峰中间。 内标法的优点是测定的结果较为准确,由于通过测量内标物及被测组分的峰面积的相对值来进行计算的,因而在一定程度上消除了操作条件等的变化所引起的误差。内标法的缺点是操作程序较为麻烦,每次分析时内标物和试样都要准确称量,有时寻找合适的内标物也有困难。 外标法简便,但进样量要求十分准确,要严格控制在与标准物相同的操作条件下进行,否则造成分析误差,得不到准确的测量结果。
内标与外标都是定量的一种方法而已,至于哪一种方法好与不好不能一概而论,做不同的分析,面对着不同的要求,再加上分析成本分析效率等等问题,我想简单而有效进行定量分析来满足要求才是最重要的。
1、以前做过很多医药、农药中间体的芳香族卤代化合物的常量定量分析,没有自动进样器,用外标法定量,确实重现性与稳定性非常差,结果经常受到搞合成同事的质疑。其实,仔细分析原因不一定就是外标法不适合这种定量分析,首先我们的实验室仪器和手段是否调整到一种稳定而合理的状态了,比如,衬管是否洁净,玻璃棉的位置是否合适恰当(能否使样品尽可能的汽化)、汽化温度是否合适、色谱峰形是否对称(也就是样品与色谱柱健合相是否匹配)、附近有没有其它色谱峰的干扰、选用什么进样方式(如快速进样还是热针进样)等等因素的影响都需要考虑,如果这些因素都考虑了,按照GMP方法验证对于精密度的要求,同一样品进6针以上的RSD和配制6个样品的定量结果RSD都能满足小于1.5%的要求,那么这个方法用外标法就是完全适用的,但是前面的影响因素是一定要都考虑到的,否则谈论这个方法是否适用就有失偏颇了。在做过的许多出口产品的定量分析方法当中有许多是一些医药公司提供的比较完善而验证过的方法,内标与外标都有(他们用的都是自动进样)精密度都能满足RSD小于1.5%的要求,当一个方法能够满足测试要求的时候,无论内标外标,都是可行的,当然有一个分析成本和分析时间的问题,内标的成本和控制溶液、样品溶液的配制当然要比外标要高和麻烦一些了。而有些时候,可能受你实验室现有仪器和附属设备的影响,达不到一定的要求,而还必须进行定量分析,有时外标的结果可能就要差一些,这时,你可能就要考虑用内标法了,可以排除手动进样的误差、分流歧视的影响、包括一些未知因素平行误差的影响,这时内标可能就显示出它的优势来了。
2、上面已经提到当做方法验证的时候,当同一样品配制6个样品溶液用所选用的外标法进行定量的时候,RSD都满足1.5%的要求时,也分为两种情况,小于1%和大于1%小于1.5%。如果RSD的结果小于1%,那这个方法就没有什么可以怀疑的了;如果RSD的结果大于1%而在1.5%略低一些的范围活动时,这个方法的可行性就将受到质疑,毕竟这是方法验证,你就要考虑上面1所提到的影响因素的影响了,如果排除掉以上的影响因素,RSD还是在1.5%附近,就要尝试内标了,如果内标结果的RSD很好,就证明你的这个方法受实验条件的影响很大,只能用内标了,或者干脆将原方法做大的变动,再尝试用外标法测试。
3、而对于微量分析,比如农药和兽药残留的分析、环境分析等,根据不同的限量标准要求对于精密度的要求也比常量分析的要求要宽松的多,RSD有时可以允许达到10%甚至更高,这时可能外标法有更大的应用空间。
4、单从精密度方面去考虑,排除其它成本和效率的因素,个人认为还是内标优于外标。曾经做过一个中间体二氨基丙醇的常量定量分析,以二乙醇胺为内标,RTX-5 amine(碱改性) 15m*0.32mm*1.0um色谱柱分析,将配制好的控制溶液(含有内标物)自动进样器进6针,目的物(二氨基丙醇)与内标物(二乙醇胺)峰面积比率的RSD为0.18%,而只对这六针样品的目的物峰(二氨基丙醇)面积求RSD,结果为0.71%,通过这一实例的结果大家就会发现到底哪个方法精密度更好了,当然是内标更好了。当然这个化合物的检测方法最后根据上面的验证数据用内标和外标定量都是可以的,实验室可以自由选择。但内标与外标精密度结果的差异是显然存在的事实。
结论:应用外标法能够满足要求,首选还是外标法了,毕竟简单而省事。对于精密度要求比较高、结果准确度会产生重大影响、实验室条件不是很理想的等等条件下,用内标法还是必要的。无论应用那种方法,方法的验证和确认都是很重要的,只要是按照程序经过验证和确认的方法,都有其应用的空间的。
外标法
用被测化合物的纯品作为标准样品,配制成一系列的已知浓度的标样。
注入色谱柱的到其响应值(峰面积)。
在一定范围内,标样的浓度与响应值之间存在较好的线性关系,即W=f×A,制成标准曲线。
在完全相同的实验条件下,注入未知样品,得到欲测组分的响应值。
根据已知的系数f,即可求出欲测组分的浓度。外标法的优点:
操作、计算简单,是一种常用的定量方法。
无需各组分都被检出、洗脱。
需要标样。
标样及未知样品的测定条件要一致。
进样体积要准确。内标法操作:
将已知量的内标样加入标准样品,制成混合标样,并配制一系列的已知浓度的工作标样。混合标样中标样与内标样的摩尔比不变。
注入色谱柱,以(标样峰面积/内标样峰面积)为响应值。
根据响应值与工作标样浓度之间存在的线性关系,即W=f×A,制成标准曲线。
将已知量的内标样加入未知样品,注入色谱柱,得到欲测组分的响应值。
根据已知的系数f,即可求出欲测组分的浓度。内标法的特点:
操作过程中样品和内标是混合在一起注入色谱柱的,因此只要混合溶液中被测组分与内标的量的比值恒定,上样体积的变化不会影响影响定量结果。
内标法抵消了上样体积,乃至流动相、检测器的影响,因此比外标法精确。 -
质谱——质谱图解析流程
lifejourney 发布于 2009-04-23 10:59:26
未知样的质谱图解析流程
(一)解析分子离子区
(1) 标出各峰的质荷比数,尤其注意高质荷比区的峰。
(2) 识别分子离子峰。首先在高质荷比区假定分子离子峰,判断该假定分子离子峰与相邻碎片离子峰关系是否合理,然后判断其是否符合氮律。若二者均相符,可认为是分子离子峰。
(3) 分析同位素峰簇的相对强度比及峰与峰间的Dm值,判断化合物是否含有C1、Br、S、Si等元素及F、P、I等无同位素的元素。
(4)推导分子式,计算不饱和度。由高分辨质谱仪测得的精确分子量或由同位素峰簇的相对强度计算分子式。若二者均难以实现时,则由分子离子峰丢失的碎片及主要碎片离子推导,或与其它方法配合。
(5)由分子离子峰的相对强度了解分子结构的信息。分子离子峰的相对强度由分子的结构所决定,结构稳定性大,相对强度就大。对于分子量约200的化合物,若分子离子峰为基峰或强蜂,谱图中碎片离子较少、表明该化合物是高稳定性分子,可能为芳烃或稠环化合物。 例如:萘分子离子峰m/z128为基峰,蒽醌分子离子峰m/z 208也是基峰。分子离子峰弱或不出现,化合物可能为多支链烃类、醇类、酸类等。
(二)、解析碎片离子
(1) 由特征离子峰及丢失的中性碎片了解可能的结构信息。 若质谱图中出现系列CnH2n+1峰,则化合物可能含长链烷基。若出现或部分出现m/z77,66,65,51,40,39等弱的碎片离子蜂,表明化合物含有苯基。若m/z91或105为基峰或强峰,表明化合物含有苄基或苯甲酰基。若质谱图中基峰或强峰出现在质荷比的中部,而其它碎片离子峰少,则化合物可能由两部分结构较稳定,其间由容易断裂的弱键相连。
(2)综合分析以上得到的全部信息,结合分子式及不饱和度,提出化合物的可能结构。
(3)分析所推导的可能结构的裂解机理,看其是否与质谱图相符,确定其结构,并进一步解释质谱,或与标准谱图比较,或与其它谱(1HNMR、13CNMR、IR)配合,确证结构。
-
标准品使用实际案例解析
颖儿飞飞 发布于 2017-08-31 11:43:10
标准品在分析检测过程中起着至关重要的作用,广泛应用于食品、农药、兽药及环境等领域,标准品的正确选择及使用关系到整个测量结果的可靠性、准确性及溯源性。根据客户反馈工作中遇到的难点和痛点,分析测试百科网旗下商城安特百货联合天津阿尔塔科技First Standard®标准品专业技术团队,推出“标准品使用实际案例解析”网络讲座,欢迎大家报名参加!
【内容要点】
1、如何选择适合自己的标准品?
2、标准溶液配制的过程中有哪些注意事项(最小取样量,溶剂选择)?
3、样品量非常少的时候如何称取?
4、如何将配制过程中产生的误差控制到最小范围?
5、混标配制有哪些难点?
6、标准品如何保存?
7、COA文件中有哪些重要信息?
8、如何选择合适的检测器?
9、衍生化反应不成功的原因有哪些?
所有参与报名实验室用户将获得由安特百货提供30元尊享红包,在线下单可抵扣现金。
点击即可免费报名:http://vote.antpedia.com/index.php?sid=785382&lang=zh-Hans
-
[论坛]webinar:LCMS在临床检测研究的全流程解决方案
颖儿飞飞 发布于 2016-07-21 17:02:46
【讲座内容】
三重串联四极杆质谱作为性能优异的检测器,具有选择性好、灵敏度高、通量大等特点,其与超高效液相色谱的联用平台,越来越多地被应用于临床实验室,作为检测复杂生物样本(血液、尿液、组织、唾液等)中多种疾病标志物的首选方案。儿茶酚胺和甲氧基肾上腺素及其在尿液和血浆中相关代谢物作为嗜铬细胞瘤的特异性标志物,近年来成为临床检测热点。
本次讲座将系统介绍安捷伦公司从生物样本前处理到分析报告的全流程解决方案,为临床研究实验室提供重要的参考策略。
注册报名:http://vote.antpedia.com/index.php?sid=758318&lang=zh-Hans
讲座时间:2016年7月26日 上午 10:00
主讲人:张政祥,2009年获北京大学理学博士学位,同年6月加入安捷伦科技(中国)有限公司,从事色谱/电泳/质谱在生物制药/制药、临床研究、组学等领域的应用支持工作,在国际期刊发表学术论文15篇,并于2013年美国质谱年会(ASMS)上获邀作oral presentation。
讲座结束后,我们将从注册并准时参会者中抽取4名幸运奖,赠送价值50元的京东礼品卡!
欲了解更多讲座信息,请加入webinar讨论群:http://www.antpedia.com/?mygroup-282 -
[论坛]最新代谢流分析软件培训讲座,使代谢研究更上一层楼!
颖儿飞飞 发布于 2016-06-27 17:41:08
【讲座内容】
“VistaFlux”为安捷伦2016年最新推出的代谢组学解决方案,整体工作流程从建立靶向代谢通路与相关代谢物数据库(PCDL),配合安捷伦MassHunter Profinder的数据提取,结合最新安捷伦OMIX软件的可视化功能可帮助相关研究人员快速且精准完成代谢流分析(Flux Analysis)。
安捷伦更利用Seahorse监控细胞能量变化与VistaFlux分析互相验证,提供科学家更深入了解细胞能量代谢通路上的变化并进一步解释更深层的生物学意义。
注册报名:http://vote.antpedia.com/index.php?sid=946968&lang=zh-Hans
讲座时间:2016年6月30日 上午 10:00
讲座详情:http://www.antpedia.com/webinar/946968.html
主讲人:詹舜安(Chan,Jimmy),安捷伦科技大中华区(华东区)LCMS应用经理。主要研究应用方向:代谢组学与多组学整合相关研究、临床分析、毒物/法医分析、食品/环境分析 等。
讲座结束后,我们将从注册并准时参会者中抽取4名幸运奖,赠送价值50元的京东礼品卡! -
容易污染液质的化合物
mass 发布于 2007-08-11 01:23:39
由Dr.John Langley,University of Southampton研究得出
(原文见: http://www.soton.ac.uk/~msweb/ )
如果有些峰一直都洗不掉,不妨找找看,在list里面有没有
Contamination Peaks - Positive Ion ES
-
[论坛]webinar:液质软件即时展示: 筛查和定量(法医应用)的全新体验
颖儿飞飞 发布于 2016-04-13 16:49:35
【内容简介】
SCIEX OS软件是为SCIEX X500R QTOF液相色谱质谱系统完全重新设计的软件。 本次网络讲座中,我们将为大家展示SCIEX OS软件,内容涵盖法医筛查和定量的数据处理。
本次讲座将包括如下要点:
1. SCIEX OS软件有一个新的直观和简化的用户界面;
2. SCIEX OS软件整合了Analyst TF, MasterViewTM, MultiQuantTM三方软件 为一体,在相同界面进行如下操作:仪器控制,数据采集,处理分析和报告;
3. SCIEX OS为用户提供了真正的下一代体验,提高效率。
注册报名:http://vote.antpedia.com/index.php?sid=413734&lang=zh-Hans
讲座时间:2016年4月15 日 上午9:30
主讲人:和翔,博士,SCIEX(美国加州红木城)资深科学家 。南京大学化学学士,美国纽约州立大学石溪分校化学博士。2012年底加入SCIEX公司,继续 临床及法医鉴定毒理学的技术开发及市场拓展。在国际SCI学术期刊(包括Analytical Chemistry, JBC, JACS, JOC, Organic Letters等)发表论文二十篇。
讲座结束后,我们将从注册并准时参会者中抽取4名幸运奖,赠送 价值50元 的京东礼品卡! -
质谱沙龙2015年学术年会
troy.tper.lee 发布于 2015-11-17 08:25:01
尊敬的各位朋友:
大家好!质谱沙龙活动从2007年开始,已经走进了第八个年头,活动在大家的亲切爱护和鼎立支持下,成功申请2013~2015年北京市继续教育项目,圆满召开“北京地区质谱沙龙年度报告交流会”。质谱沙龙一直秉承自由交流、技术共享、促进合作的宗旨,通过活动希望吸引质谱领域的专家学者互动交流,开拓质谱在医院应用研究的新视野,促进质谱技术在医药科研工作中的应用。
即将举办质谱沙龙2015年学术年会,诚挚邀请您的参与!
会议信息:
时间:2015年11月21日(周六8:30~17:30)
地点:北京朝阳医院门诊楼10层报告厅
内容:12个专家报告、讨论答疑。
其它说明:1)本活动不收取任何费用,欢迎各院校、科研单位、企业质谱技术人员及爱好者参加;2)本次活动授予Ⅰ类继续教育学分,请您携带学分卡。
邀请函详见附件,请尽量提供回执信息,方便我们安排午餐,感谢您的关注!
北京朝阳医院药事部 李鹏飞 -
质谱介绍及质谱图的解析
chongwenmen 发布于 2010-05-11 10:36:08
质谱法是将被测物质离子化,按离子的质荷比分离,测量各种离子谱峰的强度而实现分析目的的一种分析方法。质量是物质的固有特征之一,不同的物质有不同的质量谱——质谱,利用这一性质,可以进行定性分析(包括分子质量和相关结构信息);谱峰强度也与它代表的化合物含量有关,可以用于定量分析。
质谱仪一般由四部分组成:进样系统——按电离方式的需要,将样品送入离子源的适当部位;离子源——用来使样品分子电离生成离子,并使生成的离子会聚成有一定能量和几何形状的离子束;质量分析器——利用电磁场(包括磁场、磁场和电场的组合、高频电场、和高频脉冲电场等)的作用将来自离子源的离子束中不同质荷比的离子按空间位置,时间先后或运动轨道稳定与否等形式进行分离;检测器——用来接受、检测和记录被分离后的离子信号。一般情况下,进样系统将待测物在不破坏系统真空的情况下导入离子源(10-6~10-8mmHg),离子化后由质量分析器分离再检测;计算机系统对仪器进行控制、采集和处理数据,并可将质谱图与数据库中的谱图进行比较。
一、 进样系统和接口技术
将样品导入质谱仪可分为直接进样和通过接口两种方式实现。
1. 直接进样
在室温和常压下,气态或液态样品可通过一个可调喷口装置以中性流的形式导入离子源。吸附在固体上或溶解在液体中的挥发性物质可通过顶空分析器进行富集,利用吸附柱捕集,再采用程序升温的方式使之解吸,经毛细管导入质谱仪。
对于固体样品,常用进样杆直接导入。将样品置于进样杆顶部的小坩埚中,通过在离子源附近的真空环境中加热的方式导入样品,或者可通过在离子化室中将样品从一可迅速加热的金属丝上解吸或者使用激光辅助解吸的方式进行。这种方法可与电子轰击电离、化学电离以及场电离结合,适用于热稳定性差或者难挥发物的分析。
目前质谱进样系统发展较快的是多种液相色谱/质谱联用的接口技术,用以将色谱流出物导入质谱,经离子化后供质谱分析。主要技术包括各种喷雾技术(电喷雾,热喷雾和离子喷雾);传送装置(粒子束)和粒子诱导解吸(快原子轰击)等。
2. 电喷雾接口
带有样品的色谱流动相通过一个带有数千伏高压的针尖喷口喷出,生成带电液滴,经干燥气除去溶剂后,带电离子通过毛细管或者小孔直接进入质量分析器。传统的电喷雾接口只适用于流动相流速为1~5μl/min的体系,因此电喷雾接口主要适用于微柱液相色谱。同时由于离子可以带多电荷,使得高分子物质的质荷比落入大多数四极杆或磁质量分析器的分析范围(质荷比小于4000),从而可分析分子量高达几十万道尔顿(Da)的物质。
3. 热喷雾接口
存在于挥发性缓冲液流动相(如乙酸铵溶液)中的待测物,由细径管导入离子源,同时加热,溶剂在细径管中除去,待测物进入气相。其中性分子可以通过与气相中的缓冲液离子(如NH4+)反应,以化学电离的方式离子化,再被导入质量分析器。热喷雾接口适用的液体流量可达2ml/min,并适合于含有大量水的流动相,可用于测定各种极性化合物。由于在溶剂挥发时需要利用较高温度加热,因此待测物有可能受热分解。
4. 离子喷雾接口
在电喷雾接口基础上,利用气体辅助进行喷雾,可提高流动相流速达到1ml/min。电喷雾和离子喷雾技术中使用的流动相体系含有的缓冲液必须是挥发性的。
5. 粒子束接口
将色谱流出物转化为气溶胶,于脱溶剂室脱去溶剂,得到的中性待测物分子导入离子源,使用电子轰击或者化学电离的方式将其离子化,获得的质谱为经典的电子轰击电离或者化学电离质谱图,其中前者含有丰富的样品分子结构信息。但粒子束接口对样品的极性,热稳定性和分子质量有一定限制,最适用于分子量在1000Da以下的有机小分子测定。
6. 解吸附技术
将微柱液相色谱与粒子诱导解吸技术(快原子轰击,液相二次粒子质谱)结合,一般使用的流速在1~10μl/min之间,流动相须加入微量难挥发液体(如甘油)。混合液体通过一根毛细管流到置于离子源中的金属靶上,经溶剂挥发后形成的液膜被高能原子或者离子轰击而离子化。得到的质谱图与快原子轰击或者液相二次离子质谱的质谱图类似,但是本底却大大降低。
二、 离子源
离子源的性能决定了离子化效率,很大程度上决定了质谱仪的灵敏度。常见的离子化方式有两种:一种是样品在离子源中以气体的形式被离子化,另一种为从固体表面或溶液中溅射出带电离子。在很多情况下进样和离子化同时进行。
1. 电子轰击电离(EI)
气化后的样品分子进入离子化室后,受到由钨或铼灯丝发射并加速的电子流的轰击产生正离子。离子化室压力保持在 10-4~10-6mmHg。轰击电子的能量大于样品分子的电离能,使样品分子电离或碎裂。电子轰击质谱能提供有机化合物最丰富的结构信息,有较好的重现性,其裂解规律的研究也最为完善,已经建立了数万种有机化合物的标准谱图库可供检索。其缺点在于不适用于难挥发和热稳定性差的样品。
2. 化学电离(CI)
引入一定压力的反应气进入离子化室,反应气在具有一定能量的电子流的作用下电离或者裂解。生成的离子和反应气分子进一步反应或与样品分子发生离子分子反应,通过质子交换使样品分子电离。常用的反应气有甲烷,异丁烷和氨气。化学电离通常得到准分子离子,如果样品分子的质子亲和势大于反应气的质子亲和势,则生成[M+H]+,反之则生成[M-H]+。根据反应气压力不同,化学电离源分为大气压、中气压(0.1~10mmHg)和低气压(10-6mmHg)三种。大气压化学电离源适合于色谱和质谱联用,检测灵敏度较一般的化学电离源要高2~3个数量级,低气压化学电离源可以在较低的温度下分析难挥发的样品,并能使用难挥发的反应试剂,但是只能用于傅里叶变换质谱仪。
3. 快原子轰击(FAB)
将样品分散于基质(常用甘油等高沸点溶剂)制成溶液,涂布于金属靶上送入FAB离子源中。将经强电场加速后的惰性气体中性原子束(如氙)对准靶上样品轰击。基质中存在的缔合离子及经快原子轰击产生的样品离子一起被溅射进入气相,并在电场作用下进入质量分析器。如用惰性气体离子束(如铯或氩)来取代中性原子束进行轰击,所得质谱称为液相二次离子质谱(LSIMS)。
此法优点在于离子化能力强,可用于强极性、挥发性低、热稳定性差和相对分子质量大的样品及EI和CI难于得到有意义的质谱的样品。FAB比EI容易得到比较强的分子离子或准分子离子;不同于CI的一个优势在于其所得质谱有较多的碎片离子峰信息,有助于结构解析。
随着毛细管气相色谱的应用和高速真空泵的使用,现在气相色谱流出物已可直接导入质谱。
(2) 液相色谱/质谱联用(HPLC/MS)
液相色谱/质谱联用的接口前已论及,主要用于分析GC/MS不能分析,或热稳定性差,强极性和高分子量的物质,如生物样品(药物与其代谢产物)和生物大分子(肽、蛋白、核酸和多糖)。
(3) 毛细管电泳/质谱联用(CE/MS)和芯片/质谱联用(Chip/MS)
毛细管电泳(CE)适用于分离分析极微量样品(nl体积)和特定用途(如手性对映体分离等)。CE流出物可直接导入质谱,或加入辅助流动相以达到和质谱仪相匹配。微流控芯片技术是近年来发展迅速,可实现分离、过滤、衍生等多种实验室技术于一块芯片上的微型化技术,具有高通量、微型化等优点,目前也已实现芯片和质谱联用,但尚未商品化。
(4) 超临界流体色谱/质谱联用(SFC/MS)
常用超临界流体二氧化碳作流动相的SFC适用于小极性和中等极性物质的分离分析,通过色谱柱和离子源之间的分离器可实现SFC和MS联用。
(5) 等离子体发射光谱/质谱联用(ICP/MS)
由ICP作为离子源和MS 实现联用,主要用于元素分析和元素形态分析。
五、 数据处理和应用
检测器通常为光电倍增器或电子倍增器,所采集的信号经放大并转化为数字信号,计算机进行处理后得到质谱图。质谱离子的多少用丰度表示(abundance)表示,即具有某质荷比离子的数量。由于某个具体离子的“数量”无法测定,故一般用相对丰度表示其强度,即最强的峰叫基峰(base peak),其他离子的丰度用相对于基峰的百分数表示。在质谱仪测定的质量范围内,由离子的质荷比和其相对丰度构成质谱图。在LC/MS和GC/MS中,常用各分析物质的色谱保留时间和由质谱得到其离子的相对强度组成色谱总离子流图。也可确定某固定的质荷比,对整个色谱流出物进行选择离子检测(selected ion monitoring, SIM),得到选择离子流图。质谱仪分离离子的能力称为分辨率,通常定义为高度相同的相邻两峰,当两峰的峰谷高度为峰高的10%时,两峰质量的平均值与它们的质量差的比值。对于低、中、高分辨率的质谱,分别是指其分辨率在100~2000、2000~10000和10000以上。
质谱在药物领域的主要应用为药物的定性鉴别、定量分析和结构解析。
如果一个中性分子丢失或得到一个电子,则分子离子的质荷比与该分子质量数相同。使用高分辨率质谱可得到离子的精确质量数,然后计算出该化合物的分子式,或者用参照物作峰匹配可以确证分子量和分子式。分子离子的各种化学键发生断裂后形成碎片离子,由此可推断其裂解方式,得到相应的结构信息。
质谱用于定量分析,其选择性、精度和准确度较高。化合物通过直接进样或利用气相色谱和液相色谱分离纯化后再导入质谱。质谱定量分析用外标法或内标法,后者精度高于前者。定量分析中的内标可选用类似结构物质或同位素物质。前者成本低,但精度和准确度以使用同位素物质为高。使用同位素物质为内标时,要求在进样、分离和离子化过程中不会丢失同位素物质。在使用FAB质谱和LC/MS(热喷雾和电喷雾)进行定量分析时,一般都需要用稳定的同位素内标。分析物和内标离子的相对丰度采用选择离子监测(只监测分析物和内标的特定离子)的方式测定。选择离子监测相对全范围扫描而言,由于离子流积分时间长而增加了选择性和灵敏度。利用分析物和内标的色谱峰面积或峰高比得出校正曲线,然后计算样品中分析物的色谱峰面积或它的量。
解析未知样的质谱图,大致按以下程序进行。
(一)解析分子离子区
(1) 标出各峰的质荷比数,尤其注意高质荷比区的峰。
(2) 识别分子离子峰。首先在高质荷比区假定分子离子峰,判断该假定分子离子峰与相邻碎片离子峰关系是否合理,然后判断其是否符合氮律。若二者均相符,可认为是分子离子峰。
(3) 分析同位素峰簇的相对强度比及峰与峰间的Dm值,判断化合物是否含有C1、Br、S、Si等元素及F、P、I等无同位素的元素。
(4) 推导分子式,计算不饱和度。由高分辨质谱仪测得的精确分子量或由同位素峰簇的相对强度计算分子式。若二者均难以实现时,则由分子离子峰丢失的碎片及主要碎片离子推导,或与其它方法配合。
(5) 由分子离子峰的相对强度了解分子结构的信息。分子离子峰的相对强度由分子的结构所决定,结构稳定性大,相对强度就大。对于分子量约200的化合物,若分子离子峰为基峰或强蜂,谱图中碎片离子较少、表明该化合物是高稳定性分子,可能为芳烃或稠环化合物。
例如:萘分子离子峰m/z 128为基峰,蒽醌分子离子峰m/z 208也是基峰。
分子离子峰弱或不出现,化合物可能为多支链烃类、醇类、酸类等。
(二)、解析碎片离子
(1) 由特征离子峰及丢失的中性碎片了解可能的结构信息。
若质谱图中出现系列CnH2n+1峰,则化合物可能含长链烷基。若出现或部分出现m/z 77,66,65,51,40,39等弱的碎片离子蜂,表明化合物含有苯基。若m/z 91或105为基峰或强峰,表明化合物含有苄基或苯甲酰基。若质谱图中基峰或强峰出现在质荷比的中部,而其它碎片离子峰少,则化合物可能由两部分结构较稳定,其间由容易断裂的弱键相连。
(2) 综合分析以上得到的全部信息,结合分子式及不饱和度,提出化合物的可能结构。
(3) 分析所推导的可能结构的裂解机理,看其是否与质谱图相符,确定其结构,并进一步解释质谱,或与标准谱图比较,或与其它谱(1H NMR、13C NMR、IR)配合,确证结构。

