-
纯度和含量的区别和联系
色谱专家 发布于 2013-01-29 15:31:00
纯度:只表明试样中某些杂质离子存在多少的情况,即试样纯度的高低程度。纯度高,说明杂质少,并不表示试样中主体所含的量也一定高。
测定方法:测定纯度一般采用化学分析方法或仪器分析方法,先测出要求测的所有杂质含量,然后通过计算求出其纯度。
1.纯度是由100%减去杂质总量计算求得的。
2.在计算杂质总量时,通常只累计试样中存在的金属阳离子和某些非金属离子(如磷、硫、硅等)的总量,而一般不累计阴离子的总量。
3.在累计阳离子杂质总量时,不必要也不可能求出所有阳离子的杂质总量,而通常只累计常见元素、主体的伴生元素和工艺中容易引进的元素,其数目为十多个、二十多个或三十多个不等, 根据试样的情况和测试手段不同而定。
表示方法:目前可以从99.99%-99.99999%,随着测试水平的提高“9"的数目也在逐渐增加。
含量:含量的概念是表示试样中所含主体量的多少。
测定方法:含量的方法通常采用容量法和重量法等。
表示方法:由于测定方法中有效数字的限制,一般准确至小数点后两位数,一般为XX.X%-XX.XX%。
从这两种表示法和它们测定方法的不同也可看出含量与纯度之间的差别。
例如:
例1.高纯盐酸
纯度:99.9999%
含量:36~38%
阳离子:铜、铁、铅等十多项常见杂质元素的总和小于0.0001%
阴离子:略,
显而易见,在纯度为6N的高纯盐酸中含量这么低的主要原因是试样中含有大量的水。
例2.高纯铜粉
纯度:99.99%
含量:98%
阳离子:钴、镍、铅、铁等十多项常见杂质元素的总和小于0.01%
在这个标准中,铜粉含量偏低的主要因素是粉末试样在空气中容易氧化,即在试样中含有较多的氧。
从上面的例子可以看出,纯度很高的试剂的含量可能很低,反过来,含量很高(如99.9%)的试剂其纯度不一定很高。因此,在高纯度的情况下(纯度在99.99%以上)含量与纯度基本上是反映两个不同的概念。
高纯试剂的质量是否比基准试剂好?能否代替基准试剂使用?
这个问题的实质也是含量与纯度的关系问题。基准试剂与其他试剂的主要区别是试样中主体成分的含量高而准。其含量的允许范围为99.98%~100.02%(第一基准)和99.95%-100.05%(工作基准)。因将其用作标定工作基准和标定容量分析标堆溶液的标淮参考物质,故称“基准”。而纯度为4N~6N的高纯试剂,则只保证其纯度高,其含量不一定能达到基准试剂的水平,因而它不能代替基准试剂使用。同样的道理,一些人任意采用高纯金属代替国家标准中所规定的化合物来配制杂质标准溶液并认为这样更准确,是不合适的,这里就不一一论述了。 -
色谱柱
Jinyongchun 发布于 2009-09-05 20:52:10
液相色谱柱的选择、使用、维护和常见故障及排除液相色谱的柱子通常分为正相柱和反相柱。正相柱大多以硅胶为柱,或是在硅胶表面键合-CN,-NH3等官能团的键合相硅胶柱;反相柱填料主要以硅胶为基质,在其表面键合非极性的十八烷基官能团(ODS)称为C18柱,其它常用的反相柱还有C8,C4,C2和苯基柱等。另外还有离子交换柱,GPC柱,聚合物填料柱等。本文重点介绍反相色谱柱的选择和使用:
一、反相色谱柱的选择
1.柱子的PH值使用范围
反相柱优点是固定相稳定,应用广泛,可使用多种溶剂。但硅胶为基质的填料,使用时一定要注意流动相的PH范围。一般的C18柱PH值范围都在2-8,流动相的PH值小于2时,会导致键合相的水解;当PH值大于7时硅胶易溶解;经常使用缓冲液固定相要降解。一旦发生上述情况,色谱柱人口处会塌陷。同样填料各种不同牌号的色谱柱不尽相同。如果流动相PH较高或经常使用缓冲液时,建议选择PH范围大的柱子,例如戴安公司的Acclaim柱PH 2-9或Zorbax的PH 2-11. 5的柱子。
2.填料的端基封尾(或称封口)
把填料的残余硅羟基采用封口技术进行端基封尾,可改善对极性化合物的吸附或拖尾;含碳量增高了,有利于不易保留化合物的分离;填料稳定性好了,组分的保留时间重现性就好。如果待分析的样品属酸性或碱性的化合物,最好选用填料经端基封尾的色谱柱。
3.戴安公司Acclaim柱子介绍—极性封尾C16固定相柱
戴安公司有28种类型的柱子,Acclaim反相柱填料高纯,金属含量极低,完全封尾。PH 2-9范围内兼容,低流失,高柱效。尤其是2003年推出的Acclaim极性封尾C16柱,是最先商品化的磺酰氨-O链接键的色谱柱,具极低的硅羟基活性,能在极性溶剂甚至100%水的条件下长期使用。对酸性和碱性化合物有极为尖锐的好的色谱峰形,与现有的一流色谱柱相比有更好的立体选择性。(下图是Acclaim极性封尾C16柱和市售极性封尾一流色谱柱分离酸性化合物谱图的比较)
二、液相色谱柱的使用
色谱柱在使用前,最好进行柱的性能测试,并将结果保存起来,作为今后评价柱性能变化的参考。在做柱性能测试时要按照色谱柱出厂报告中的条件进行(出厂测试所使用的条件是最佳条件),只有这样,测得的结果才有可比性。但要注意:柱性能可能由于所使用的样品、流动相、柱温等条件的差异而有所不同。1、样品的前处理
a、最好使用流动相溶解样品。b、使用预处理柱除去样品中的强极性或与柱填料产生不 可逆吸附的杂质。
c、使用0.45µm的过滤膜过滤除去微粒杂质。
2、流动相的配制
液相色谱是样品组分在柱填料与流动相之间质量交换而达到分离的目的,因此要求流动相具备以下的特点:
a、流动相对样品具有一定的溶解能力,保证样品组分不会沉淀在柱中(或长时间保留在柱中)。
b、流动相与样品不产生化学反应
c、流动相的黏度要尽量小,以便得到好的分离效果;降低柱压降,延长泵的使用寿命(可运用提高温度的方法降低流动相的黏度)。
d、流动相的物化性质要与使用的检测器相适应。如使用UV检测器,最好使用对紫外吸收较低的溶剂配制。
e、流动相沸点不要太低,否则容易产生气泡,导致实验无法进行。f、在流动相配制好后,一定要进行脱气。除去溶解在流动相中的微量气体既有利于检测,还可以防止流动相中的微量氧与样品发生作用。
3、流动相流速的选择因柱效是柱中流动相线性流速的函数,使用不同的流速可得到不同的柱效。对于一根特定的色谱柱,要追求最佳柱效,最好使用最佳流速。对内径为4.6mm的色谱柱,流速一般选择1ml/min,对于内径为4.0mm柱,流速0.8ml/min为佳。
当选用最佳流速时,分析时间可能延长。可采用改变流动相的洗涤强度的方法以缩短分析时间(如使用反相柱时,可适当增加甲醇或乙腈的含量)。
注意:
a.含水流动相最好在实验前配制,尤其是夏天使用缓冲 溶液作为流动相不要过夜。最好加入叠氮化钠,防止细菌生长。
b.流动相要求使用0.45 µm滤膜过滤,除去微粒杂质。
c.使用HPLC级溶剂配制流动相,使用合适的流动相可延长色谱柱的使用寿命,提高柱性能。三. 色谱柱的维护
1. 色谱柱的平衡
反相色谱柱由工厂测试后是保存在乙腈/水中的。新柱应先使用10-20倍柱体积的甲醇或乙腈冲洗色谱柱。请一定确保您分析样品所使用的流动相和乙腈/水互溶。 每天用足够的时间以流动相来平衡色谱柱,您就会在处理问题方面获得最大的"补偿",而且您的色谱柱的寿命也会变得更长!操作步骤:
a. 平衡开始时将流速缓慢地提高,用流动相平衡色谱柱直到获得稳定的基线(缓冲盐或离子对试剂流速如果较低,则需要较长的时间来平衡)
b. 如果使用的流动相中含有缓冲盐,应注意用纯水"过渡"即每天分析开始前必须先用纯水冲洗30分钟以上再用缓冲盐流动相平衡; 分析结束后必须先用纯水冲洗30分钟以上除去缓冲盐之后再用甲醇冲洗30分钟保护柱子。
2. 色谱柱的再生
长期使用的色谱柱,往往柱效会下降(柱子的理论塔板数减低)。可以对色谱柱进行再生,在有条件的实验室应使用一个廉价的泵进行柱子的再生。
建议用来冲洗柱子的溶剂体积
色谱柱尺寸 柱体积 所用溶剂的体积
125-4mm 1.6ml 30ml
250-4 mm 3.2ml 60ml
250-10mm 20ml 400ml
选择再生方法:
极性固定相(如Si,NH2* ,DIOL基色谱填料)的再生:
正庚烷→氯仿→乙酸乙酯→丙酮→乙醇→水**
非极性固定相(如反相色谱填料RP-18,RP-8,CN等)的再生: 水→乙腈→氯仿(或异丙醇)→乙腈→水
注意:
a. 在对NH2改性的色谱柱进行再生时,由于NH2可能以铵根离子的形式存在,因此应该在水洗后用0.1M的氨水冲洗,然后再用水冲洗至碱溶液完全流出。
b. 0.05M稀硫酸可以用来清洗已污染的色谱柱,如果简单的用有机溶剂/水的处理不能够完全洗去硅胶表面吸附的杂质,在水洗后加用0.05M稀硫酸冲洗非常有效。3. 色谱柱的维护
a.使用预柱保护分析柱(硅胶在极性流动相/离子性流动相中有一定的溶解度)
b.大多数反相色谱柱的pH稳定范围是2-7.5,尽量不超过该色谱柱的pH范围
c.避免流动相组成及极性的剧烈变化
d.流动相使用前必须经脱气和过滤处理
e.如果使用极性或离子性的缓冲溶液作流动相,应在实验完毕柱子冲洗干净,并保存于甲醇或乙腈中
f.氯化物的溶剂对其有一定的腐蚀性,故使用时要注意,柱及连接管内不能长时间存留此类溶剂,以避免腐蚀。 -
X射线衍射分析
Chloe 发布于 2014-05-05 10:36:23
X射线衍射分析(X-ray diffraction,简称XRD),是利用晶体形成的X射线衍射,对物质进行内部原子在空间分布状况的结构分析方法。将具有一定波长的X射线照射到结晶性物质上时,X射线因在结晶内遇到规则排列的原子或离子而发生散射,散射的X射线在某些方向上相位得到加强,从而显示与结晶结构相对应的特有的衍射现象。X射线衍射方法具有不损伤样品、无污染、快捷、测量精度高、能得到有关晶体完整性的大量信息等优点。
X射线衍射分析是利用晶体形成的X射线衍射,对物质进行内部原子在空间分布状况的结构分析方法。将具有一定波长的X射线照射到结晶性物质上时,X射线因在结晶内遇到规则排列的原子或离子而发生散射,散射的X射线在某些方向上相位得到加强,从而显示与结晶结构相对应的特有的衍射现象。衍射X射线满足布拉格(W.L.Bragg)方程:2d
X射线衍射的产生
sinθ=nλ式中:λ是X射线的波长;θ是衍射角;d是结晶面间隔;n是整数。波长λ可用已知的X射线衍射角测定,进而求得面间隔,即结晶内原子或离子的规则排列状态。将求出的衍射X射线强度和面间隔与已知的表对照,即可确定试样结晶的物质结构,此即定性分析。从衍射X射线强度的比较,可进行定量分析。本法的特点在于可以获得元素存在的化合物状态、原子间相互结合的方式,从而可进行价态分析,可用于对环境固体污染物的物相鉴定,如大气颗粒物中的风砂和土壤成分、工业排放的金属及其化合物(粉尘)、汽车排气中卤化铅的组成、水体沉积物或悬浮物中金属存在的状态等等。
理论发展
1912年劳埃等人根据理论预见,并用实验证实了X射线与晶体相遇时能发生衍射现象,证明了X射线具有电磁波的性质,成为X射线衍射学的第一个里程碑。当一束单色X射线入射到晶体时,由于晶体是由原子规则排列成的晶胞组成,这些规则排列的原子间距离与入射X射线波长有相同数量级,故由不同原子散射的X射线相互干涉,在某些特殊方向上产生强X射线衍射,衍射线在空间分布的方位和强度,与晶体结构密切相关。这就是X射线衍射的基本原理。衍射线空间方位与晶体结构的关系可用布拉格方程表示:
2dsinθ=nλ
式中:λ是X射线的波长;θ是衍射角;d是结晶面间隔;n是整数。波长λ可用已知的X射线衍射角测定,进而求得面间隔,即结晶内原子或离子的规则排列状态。将求出的衍射X射线强度和面间隔与已知的表对照,即可确定试样结晶的物质结构,此即定性分析。从衍射X射线强度的比较,可进行定量分析。
运动学衍射理论
Darwin的理论称为X射线衍射运动学理论。该理论把衍射现象作为三维Fraunhofer衍射问题来处理,认为晶体的每个体积元的散射与其它体积元的散射无关,而且散射线通过晶体时不会再被散射。虽然这样处理可以得出足够精确的衍射方向,也能得出衍射强度,但运动学理论的根本性假设并不完全合理。因为散射线在晶体内一定会被再次散射,除了与原射线相结合外,散射线之间也能相互结合。Darwin不久以后就认识到这点,并在他的理论中作出了多重散射修正。
动力学衍射理论
Ewald的理论称为动力学理论。该理论考虑到了晶体内所有波的相互作用,认为入射线与衍射线在晶体内相干地结合,而且能来回地交换能量。两种理论对细小的晶体粉末得到的强度公式相同,而对大块完整的晶体,则必须采用动力学理论才能得出正确的结果。
发展方向
X射线分析的新发展,金属X射线分析由于设备和技术的普及已逐步变成金属研究和有机材料,纳米材料测试的常规方法。而且还用于动态测量。早期多用照相法,这种方法费时较长,强度测量的精确度低。50年代初问世的计数器衍射仪法具有快速、强度测量准确,并可配备计算机控制等优点,已经得到广泛的应用。但使用单色器的照相法在微量样品和探索未知新相的分析中仍有自己的特色。从70年代以来,随着高强度X射线源(包括超高强度的旋转阳极X射线发生器、电子同步加速辐射,高压脉冲X射线源)和高灵敏度探测器的出现以及电子计算机分析的应用,使金属 X射线学获得新的推动力。这些新技术的结合,不仅大大加快分析速度,提高精度,而且可以进行瞬时的动态观察以及对更为微弱或精细效应的研究。
详细内容
概述
研究晶体材料,X射线衍射方法非常理想非常有效,而对于液体和非晶态物固体,这种方法也能提供许多基本的重要数据。所以X射线衍射法被认为是研究固体最有效的工具。在各种衍射实验方法中,基本方法有单晶法、多晶法和双晶法。
单晶衍射法
单晶X射线衍射分析的基本方法为劳埃法与周转晶体法。
劳埃法
劳埃法以光源发出连续X射线照射置于样品台上静止的单晶体样品,用平板底片记录产生的衍射线。根据底片位置的不同,劳埃法可以分为透射劳埃法和背射劳埃法。背射劳埃法不受样品厚度和吸收的限制,是常用的方法。劳埃法的衍射花样由若干劳埃斑组成,每一个劳埃斑相应于晶面的1~n级反射,各劳埃斑的分布构成一条晶带曲线。
周转晶体法
周转晶体法以单色X射线照射转动的单晶样品,用以样品转动轴为轴线的圆柱形底片记录产生的衍射线,在底片上形成分立的衍射斑。这样的衍射花样容易准确测定晶体的衍射方向和衍射强度,适用于未知晶体的结构分析。周转晶体法很容易分析对称性较低的晶体(如正交、单斜、三斜等晶系晶体)结构,但应用较少。
多晶衍射法
多晶X射线衍射方法包括照相法与衍射仪法。
照相法
照相法以光源发出的特征X射线照射多晶样品,并用底片记录衍射花样。根据样品与底片的相对位置,照相法可以分为德拜法、聚焦法和针孔法,其中德拜法应用最为普遍。
德拜法以一束准直的特征X射线照射到小块粉末样品上,用卷成圆柱状并与样品同轴安装的窄条底片记录衍射信息,获得的衍射花样是一些衍射弧。此方法的优点为:⑴ 所用试样量少(0.1毫克即可);⑵ 包含了试样产生的全部反射线;⑶ 装置和技术比较简单。
聚焦法的底片与样品处于同一圆周上,以具有较大发散度的单色X射线照射样品上较大区域。由于同一圆周上的同弧圆周角相等,使得多晶样品中的等同晶面的衍射线在底片上聚焦成一点或一条线。聚焦法曝光时间短,分辨率是德拜法的两倍,但在小θ 范围衍射线条较少且宽,不适于分析未知样品。
针孔法用三个针孔准直的单色X射线为光源,照射到平板样品上。根据底片不同的位置针孔法又分为穿透针孔法和背射针孔法。针孔法得到的衍射花样是衍射线的整个圆环,适于研究晶粒大小、晶体完整性、宏观残余应力及多晶试样中的择优取向等。但这种方法只能记录很少的几个衍射环,不适于其它应用。
衍射仪法
X射线衍射仪以布拉格实验装置为原型,融合了机械与电子技术等多方面的成果。衍射仪由X射线发生器、X射线测角仪、辐射探测器和辐射探测电路4个基本部分组成,是以特征X射线照射多晶体样品,并以辐射探测器记录衍射信息的衍射实验装置。现代X射线衍射仪还配有控制操作和运行软件的计算机系统。X射线衍射仪的成像原理与聚集法相同,但记录方式及相应获得的衍射花样不同。衍射仪采用具有一定发散度的入射线,也用“同一圆周上的同弧圆周角相等”的原理聚焦,不同的是其聚焦圆半径随 2θ的变化而变化。衍射仪法以其方便、快捷、准确和可以自动进行数据处理等特点在许多领域中取代了照相法,现在已成为晶体结构分析等工作的主要方法。
双晶衍射法
双晶衍射仪用一束X射线(通常用Ka1作为射线源)照射一个参考晶体的表面,使符合布拉格条件的某一波长的X射线在很小角度范围内被反射,这样便得到接近单色并受到偏振化的窄反射线,再用适当的光阑作为限制,就得到近乎准值的X射线束。把此X射线作为第二晶体的入射线,第二晶体和计数管在衍射位置附近分别以Δθ 及Δ(2θ)角度摆动,就形成通常的双晶衍射仪。
在近完整晶体中,缺陷、畸变等体现在X射线谱中只有几十弧秒,而半导体材料进行外延生长要求晶格失配要达到10-4或更小。这样精细的要求使双晶X射线衍射技术成为近代光电子材料及器件研制的必备测量仪器,以双晶衍射技术为基础而发展起来的四晶及五晶衍射技术(亦称为双晶衍射),已成为近代X射线衍射技术取得突出成就的标志。但双晶衍射仪的第二晶体最好与第一晶体是同种晶体,否则会发生色散。所以在测量时,双晶衍射仪的参考晶体要与被测晶体相同,这个要求使双晶衍射仪的使用受到限制。
-
X射线衍射法
Chloe 发布于 2014-05-05 10:43:08
概念
X射线晶体学是一门利用X射线来研究晶体中原子排列的学科。更准确地说,利用电子对X射线的散射作用,X射线晶体学可以获得晶体中电子密度的分布情况,再从中分析获得原子的位置信息,即晶体结构。(以下论述以高分子材料的X射线晶体学为主)由于所有的原子都含有电子,并且X射线的波长范围为0.001-10纳米(即0.01-100埃),其波长与成键原子之间的距离(1-2埃附近)可比,因此X射线可用于研究各类分子的结构。但是,到目前为止还不能用X射线对单个的分子成像,因为没有X射线透镜可以聚焦X射线,而且X射线对单个分子的衍射能力非常弱,无法被探测。而晶体(一般为单晶)中含有数量巨大的方位相同的分子,X射线对这些分子的衍射叠加在一起就能够产生足以被探测的信号。从这个意义上说,晶体就是一个X射线的信号放大器。X射线晶体学将X射线与晶体学联系在一起,从而可以对各类晶体结构进行研究,特别是蛋白质晶体结构。
根据X射线穿过物质的晶格时所产生的衍射特征,鉴定物质成分与结构的方法。利用晶体对X射线的衍射效应,研究晶体的内部结构,最终确定出不同的或相同的原子在晶胞内的位置(即原子的排列方式)。它包括:①根据晶体的晶形、劳埃图以及某些物理性质(如压电性、旋光性等),确定出晶体的晶系和对称型;②根据回摆图或旋转图测定出晶胞参数;③根据晶体化学组成及其密度和晶胞参数,计算出单位晶胞内分子数,从而算出单位晶胞内各种原子的数目;④对魏森堡图或回摆图进行指标化,即对照片上每一衍射点确定其晶面指标的过程,然后根据衍射系统消光的特点定出衍射群,再结合其他性质定出空间群;⑤根据衍射点的指标和对应每一衍射点的衍射强度,并通过对强度数据进行一系列修正,还原为结构振幅;⑥再根据这许多由实验得到的结构振幅资料,或运用直接法(求出其相角),或结合晶体化学原理运用试差法(反复假设试用结构),最终确定出每个原子在单位晶胞内的坐标。X射线衍射技术已经成为研究粘土矿物(尤其是研究泥岩和碳酸盐岩中的粘土矿物)的最重要手段,并且也是研究各种自生矿物的重要手段,并能对泥、页岩中自生矿物和碎屑矿物做定量分析,此外还可以用于固体有机质的显微结构与变质程度的研究,这些都是其他分析手段的弱点。
X射线衍射法是一种研究晶体结构的分析方法,而不是直接研究试样内含有元素的种类及含量的方法。当X射线照射晶态结构时,将受到晶体点阵排列的不同原子或分子所衍射。X射线照射两个晶面距为d的晶面时,受到晶面的反射,两束反射X光程差2dsinθ使入射波长的整数倍时,即2dsinθ=nλ(n为整数),两束光的相位一致,发生相长干涉,这种干涉现象称为衍射,晶体对X射线的这种折射规则称为布拉格规则。θ称为衍射角(入射或衍射X射线与晶面间夹角)。n相当于相干波之间的位相差,n=1,2…时各称0级、1级、2级……衍射线。反射级次不清楚时,均以n=1求d。晶面间距一般为物质的特有参数,对一个物质若能测定数个d及与其相对应的衍射线的相对强度,则能对物质进行鉴定。
-
激光粒度仪及其原理介绍
release 发布于 2013-01-17 23:39:06
激光粒度分析仪仪是根据光的散射原理测量粉颗粒大小的,是一种比较通用的粒度仪。其特点是测量的动态范围宽、测量速度快、操作方便,尤其适合测量粒度分布范围宽的粉体和液体雾滴。对粒度均匀的粉体,比如磨料微粉,要慎重选用。激光粒度仪集成了激光技术、现代光电技术、电子技术、精密机械和计算机技术,具有测量速度快、动态范围大、操作简便、重复性好等优点,现已成为全世界最流行的粒度测试仪器。激光粒度仪作为一种新型的粒度测试仪器,已经在其它粉体加工与应用领域得到广泛的应用。它的特点是测试速度快、重复性好、准确性好、操作简便。对提高产品质量、降低能源消耗有着重要的意义。激光粒度仪的原理激光粒度仪是根据颗粒能使激光产生散射这一物理现象测试粒度分布的。由于激光具有很好的单色性和极强的方向性,所以在没有阻碍的无限空间中激光将会照射到无穷远的地方,并且在传播过程中很少有发散的现象。如图1所示。 图1 激光束在无阻碍状态下的传播示意图米氏散射理论表明,当光束遇到颗粒阻挡时,一部分光将发生散射现象,散射光的传播方向将与主光束的传播方向形成一个夹角θ,θ角的大小与颗粒的大小有关,颗粒越大,产生的散射光的θ角就越小;颗粒越小,产生的散射光的θ角就越大。即小角度(θ)的散射光是有大颗粒引起的;大角度(θ1)的散射光是由小颗粒引起的,如图2所示。进一步研究表明,散射光的强度代表该粒径颗粒的数量。这样,测量不同角度上的散射光的强度,就可以得到样品的粒度分布了。
图1 激光束在无阻碍状态下的传播示意图米氏散射理论表明,当光束遇到颗粒阻挡时,一部分光将发生散射现象,散射光的传播方向将与主光束的传播方向形成一个夹角θ,θ角的大小与颗粒的大小有关,颗粒越大,产生的散射光的θ角就越小;颗粒越小,产生的散射光的θ角就越大。即小角度(θ)的散射光是有大颗粒引起的;大角度(θ1)的散射光是由小颗粒引起的,如图2所示。进一步研究表明,散射光的强度代表该粒径颗粒的数量。这样,测量不同角度上的散射光的强度,就可以得到样品的粒度分布了。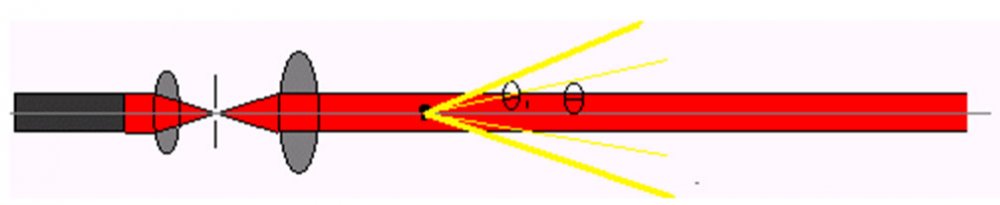 图2 不同粒径的颗粒产生不同角度的散射光为了测量不同角度上的散射光的光强,需要运用光学手段对散射光进行处理。我们在光束中的适当的位置上放置一个富氏透镜,在该富氏透镜的后焦平面上放置一组多元光电探测器,不同角度的散射光通过富氏透镜照射到多元光电探测器上时,光信号将被转换成电信号并传输到电脑中,通过专用软件对这些信号进行处理,就会准确地得到粒度分布了,如图3所示。
图2 不同粒径的颗粒产生不同角度的散射光为了测量不同角度上的散射光的光强,需要运用光学手段对散射光进行处理。我们在光束中的适当的位置上放置一个富氏透镜,在该富氏透镜的后焦平面上放置一组多元光电探测器,不同角度的散射光通过富氏透镜照射到多元光电探测器上时,光信号将被转换成电信号并传输到电脑中,通过专用软件对这些信号进行处理,就会准确地得到粒度分布了,如图3所示。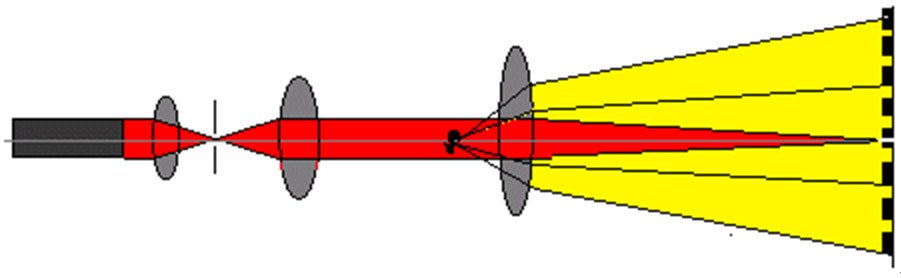 图3 激光粒度仪原理示意图激光粒度仪是根据颗粒能使激光产生散射这一物理现象测试粒度分布的。由于激光具有很好的单色性和极强的方向性,所以一束平行的激光在没有阻碍的无限空间中将会照射到无限远的地方,并且在传播过程中很少有发散的现象。如图7所示。
图3 激光粒度仪原理示意图激光粒度仪是根据颗粒能使激光产生散射这一物理现象测试粒度分布的。由于激光具有很好的单色性和极强的方向性,所以一束平行的激光在没有阻碍的无限空间中将会照射到无限远的地方,并且在传播过程中很少有发散的现象。如图7所示。 当光束遇到颗粒阻挡时,一部分光将发生散射现象,如图8。散射光的传播方向将与主光束的传播方向形成一个夹角θ。散射理论和实验结果都告诉我们,散射角θ的大小与颗粒的大小有关,颗粒越大,产生的散射光的θ角就越小;颗粒越小,产生的散射光的θ角就越大。在图8中,散射光I1是由较大颗粒引起的;散射光I2是由较小颗粒引起的。进一步研究表明,散射光的强度代表该粒径颗粒的数量。这样,在不同的角度上测量散射光的强度,就可以得到样品的粒度分布了。
当光束遇到颗粒阻挡时,一部分光将发生散射现象,如图8。散射光的传播方向将与主光束的传播方向形成一个夹角θ。散射理论和实验结果都告诉我们,散射角θ的大小与颗粒的大小有关,颗粒越大,产生的散射光的θ角就越小;颗粒越小,产生的散射光的θ角就越大。在图8中,散射光I1是由较大颗粒引起的;散射光I2是由较小颗粒引起的。进一步研究表明,散射光的强度代表该粒径颗粒的数量。这样,在不同的角度上测量散射光的强度,就可以得到样品的粒度分布了。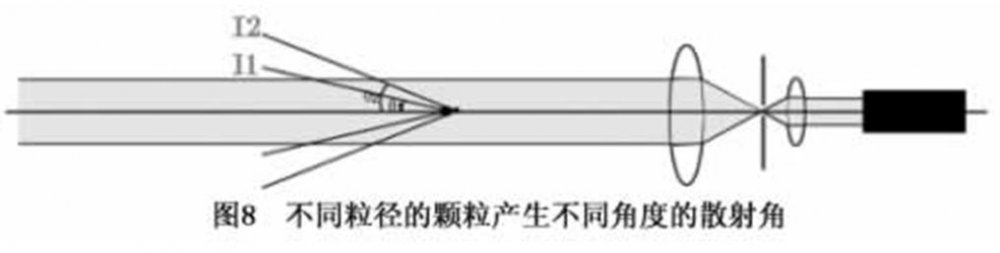 为了有效地测量不同角度上的散射光的光强,需要运用光学手段对散射光进行处理。我们在图8所示的光束中的适当的位置上放置一个富氏透镜,在该富氏透镜的后焦平面上放置一组多元光电探测器,这样不同角度的散射光通过富氏透镜就会照射到多元光电探测器上,将这些包含粒度分布信息的光信号转换成电信号并传输到电脑中,通过专用软件用Mie散射理论对这些信号进行处理,就会准确地得到所测试样品的粒度分布了,如图9所示。
为了有效地测量不同角度上的散射光的光强,需要运用光学手段对散射光进行处理。我们在图8所示的光束中的适当的位置上放置一个富氏透镜,在该富氏透镜的后焦平面上放置一组多元光电探测器,这样不同角度的散射光通过富氏透镜就会照射到多元光电探测器上,将这些包含粒度分布信息的光信号转换成电信号并传输到电脑中,通过专用软件用Mie散射理论对这些信号进行处理,就会准确地得到所测试样品的粒度分布了,如图9所示。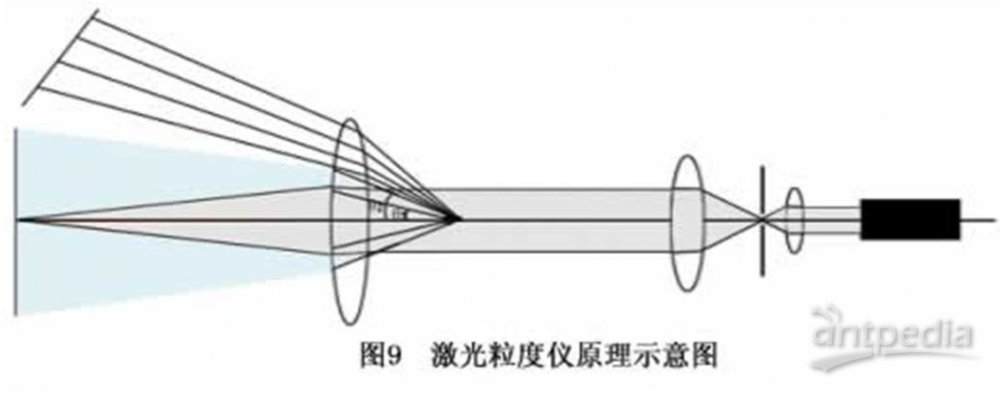 激光粒度分析仪原理光在传播中,波前受到与波长尺度相当的隙孔或颗粒的限制,以受限波前处各元波为源的发射在空间干涉而产生衍射和散射,衍射和散射的光能的空间(角度)分布与光波波长和隙孔或颗粒的尺度有关。用激光做光源,光为波长一定的单色光后,衍射和散射的光能的空间(角度)分布就只与粒径有关。对颗粒群的衍射,各颗粒级的多少决定着对应各特定角处获得的光能量的大小,各特定角光能量在总光能量中的比例,应反映着各颗粒级的分布丰度。按照这一思路可建立表征粒度级丰度与各特定角处获取的光能量的数学物理模型,进而研制仪器,测量光能,由特定角度测得的光能与总光能的比较推出颗粒群相应粒径级的丰度比例量。激光粒度分析仪采用湿法分散技术,机械搅拌使样品均匀散开,超声高频震荡使团聚的颗粒充分分散,电磁循环泵使大小颗粒在整个循环系统中均匀分布,从而在根本上保证了宽分布样品测试的准确重复。测试操作简便快捷:放入分散介质和被测样品,启动超生发生器使样品充分分散,然后启动循环泵,实际的测试过程只有几秒钟。测试结果以粒度分布数据表、分布曲线、比表面积、D10、D50、D90等方式显示、打印和记录.输出数据丰富直观:本仪器的软件可以在各种计算机视窗平台上运行,具有操作简单直观的特点,不仅对样品进行动态检测,而且具有强大的数据处理与输出功能,用户可以选择和设计最理想的表格和图形输出。
激光粒度分析仪原理光在传播中,波前受到与波长尺度相当的隙孔或颗粒的限制,以受限波前处各元波为源的发射在空间干涉而产生衍射和散射,衍射和散射的光能的空间(角度)分布与光波波长和隙孔或颗粒的尺度有关。用激光做光源,光为波长一定的单色光后,衍射和散射的光能的空间(角度)分布就只与粒径有关。对颗粒群的衍射,各颗粒级的多少决定着对应各特定角处获得的光能量的大小,各特定角光能量在总光能量中的比例,应反映着各颗粒级的分布丰度。按照这一思路可建立表征粒度级丰度与各特定角处获取的光能量的数学物理模型,进而研制仪器,测量光能,由特定角度测得的光能与总光能的比较推出颗粒群相应粒径级的丰度比例量。激光粒度分析仪采用湿法分散技术,机械搅拌使样品均匀散开,超声高频震荡使团聚的颗粒充分分散,电磁循环泵使大小颗粒在整个循环系统中均匀分布,从而在根本上保证了宽分布样品测试的准确重复。测试操作简便快捷:放入分散介质和被测样品,启动超生发生器使样品充分分散,然后启动循环泵,实际的测试过程只有几秒钟。测试结果以粒度分布数据表、分布曲线、比表面积、D10、D50、D90等方式显示、打印和记录.输出数据丰富直观:本仪器的软件可以在各种计算机视窗平台上运行,具有操作简单直观的特点,不仅对样品进行动态检测,而且具有强大的数据处理与输出功能,用户可以选择和设计最理想的表格和图形输出。 -
复合玻璃电极的存放
samgong 发布于 2010-02-11 23:59:09
酸碱滴定用复合玻璃电极分成两种,水相复合玻璃电极和非水相复合玻璃电极,电极的性能和寿命与日常测量的样品和维护有很大的关系。通常,客户拿到购买的电极后,需要立刻检查电极的外观和测量相应的性能(如进行校验)。如果发现有任何问题需要及时向销售商提出。在确认电极性能良好后,需要把电极存储在相应的溶液中。其中:水相复合玻璃电极的存储方法a. 电极填充液为3M KCl 的电极,应保存在电极存储液中(Storage solution)6.2323.000。电极存储液可以阻止电极玻璃膜的老化。也就是说,电极长时间存放后,电极的响应时间不会改变,电极每次从存储液中拿出来都可以直接使用。b. 电极填充液为其他电解质溶液时,应该保存在相应的电解质溶液里c. 电极保存时要把电极浸入到相应的溶液中,关上溶液填充口,确保玻璃隔膜完全浸没在溶液中。千万不能使电极干燥。非水相复合玻璃电极的存储方法适用于非水相的酸碱滴定电极 货号:6.0229.100非水相复合玻璃电极随机的填充液为LiCl 的乙醇饱和溶液,然而,在某些情况下(尤其是PH 大于16 时),需要其他的内参比溶液。当用碱溶液做滴定剂时,内参比溶液选用c(TEABr)=0.4mol/L的乙烯基乙二醇溶液。(TEABr 为四乙基溴化铵)当用酸溶液做滴定剂时,内参比溶液推荐c(LiCl)=2mol/L 的乙醇溶液。非水相复合玻璃电极都应保存在相应的内参比溶液中。LiCl 饱和乙醇溶液的订货号为:6.2312.0002mol/L 的LiCl 的订货号为:6.2312.010c(TEABr)=0.4mol/L 的订货号为:6.2320.000 -
离子色谱基础
ichrom 发布于 2009-09-12 12:34:16
离子色谱 (Ion Chromatography)是高效液相色谱(HPLC)的一种,是分析阴离子和阳离子的一种液相色谱方法。
一、离子色谱的基本原理
离子色谱的分离机理主要是离子交换,有3种分离方式,它们是高效离子交换色谱(HPIC)、离子排斥色谱 (HPIEC)和离子对色谱 (MPIC)。用于3种分离方式的柱填料的树脂骨架基本都是苯乙烯-二乙烯基苯的共聚物,但树脂的离子交换功能基和容量各不相同。HPIC用低容量的离子交换树脂,HPIEC用高容量的树脂,MPIC用不含离子交换基团的多孔树脂。3种分离方式各基于不同分离机理。HPIC的分离机理主要是离子交换,HPIEC主要为离子排斥,而MPIC则是主要基于吸附和离子对的形成。
1、高效离子交换色谱
应用离子交换的原理,采用低交换容量的离子交换树脂来分离离子,这在离子色谱中应用最广泛,其主要填料类型为有机离子交换树脂,以苯乙烯二乙烯苯共聚体为骨架,在苯环上引入磺酸基,形成强酸型阳离子交换树脂,引入叔胺基而成季胺型强碱性阴离子交换树脂,此交换树脂具有大孔或薄壳型或多孔表面层型的物理结构,以便于快速达到交换平衡,离子交换树脂耐酸碱可在任何pH范围内使用,易再生处理、使用寿命长,缺点是机械强度差、易溶胀易、受有机物污染。
硅质键合离子交换剂以硅胶为载体,将有离子交换基的有机硅烷与基表面的硅醇基反应,形成化学键合型离子交换剂,其特点是柱效高、交换平衡快、机械强度高,缺点是不耐酸碱、只宜在pH28范围内使用。
离子交换色谱是最常用的离子色谱。
2、离子排斥色谱
它主要根据Donnon膜排斥效应,电离组分受排斥不被保留,而弱酸则有一定保留的原理,制成离子排斥色谱主要用于分离有机酸以及无机含氧酸根,如硼酸根碳酸根和硫酸根有机酸等。它主要采用高交换容量的磺化H型阳离子交换树脂为填料以稀盐酸为淋洗液。
3、离子对色谱
离子对色谱的固定相为疏水型的中性填料,可用苯乙烯二乙烯苯树脂或十八烷基硅胶(ODS),也有用C8硅胶或CN,固定相流动相由含有所谓对离子试剂和含适量有机溶剂的水溶液组成,对离子是指其电荷与待测离子相反,并能与之生成疏水性离子,对化合物的表面活性剂离子,用于阴离子分离的对离子是烷基胺类如氢氧化四丁基铵氢氧化十六烷基三甲烷等,用于阳离子分离的对离子是烷基磺酸类,如己烷磺酸钠,庚烷磺酸钠等对离子的非极性端亲脂极性端亲水,其CH2键越长则离子对化合物在固定相的保留越强,在极性流动相中,往往加入一些有机溶剂,以加快淋洗速度,此法主要用于疏水性阴离子以及金属络合物的分离,至于其分离机理则有3种不同的假说,反相离子对分配离子交换以及离子相互作用。
二、离子色谱系统
IC系统的构成与HPLC相同,仪器由流动相传送部分、分离柱、检测器和数据处理4个部分组成,在需要抑制背景电导的情况下通常还配有MSM或类似抑制器。其主要不同之处是IC的流动相要求耐酸碱腐蚀以及在可与水互溶的有机溶剂(如乙腈、甲醇和丙酮等)中不溶胀的系统。因此,凡是流动相通过的管道、阀门、泵、柱子及接头等均不宜用不锈钢材料,而是用耐酸碱腐蚀的PEEK材料的全塑IC系统。离子色谱的最重要的部件是分离柱。柱管材料应是惰性的,一般均在室温下使用。高效柱和特殊性能分离柱的研制成功,是离子色谱迅速发展的关键。
三、离子色谱的优点
1、快速、方便:对7种常见阴离子(F-、Cl-、Br-、NO2-、NO3-、SO42-、PO43-)和6种常见阳离子(Li+、Na+、NH4+、K+、Mg2+、Ca2+)的平均分析时间已分别小于8min。用高效快速分离柱对上述7种最重要的常见阴离子达基线分离只需3min。
2、灵敏度高:离子色谱分析的浓度范围为低μg/L(1~10μg/L)至数百mg/L。直接进样(25μL),电导检测,对常见阴离子的检出限小于10μg/L。
3、选择性好:IC法分析无机和有机阴、阳离子的选择性可通过选择恰当的分离方式、分离祝贺监测方法来达到。与HPLC相比,IC中固定相对选择性的影响较大。
4、可同时分析多种离子化合物:与光度法、原子吸收法相比,IC的主要优点是可同时检测样品中的多种成分。只需很短的时间就可得到阴、阳离子以及样品组成的全部信息。
5、分离柱的稳定性好、容量高:与HPLC中所用的硅胶填料不同,IC柱填料的高pH值稳定性允许用强酸或强碱作淋洗液,有利于扩大应用范围。
四、离子色谱的检测方法
离子色谱的检测器分为两大类,即电化学检测器和光学检测器。电化学检测器包括电导、直流安培、脉冲安培和积分安培;光化学检测器包括紫外-可见和荧光。
随着离子色谱的广泛应用,离子色谱的检测技术已由单一的化学抑制型电导法发展为包括电化学光化学和与其他多种分析仪器联用的方法。 1、抑制电导检测法;2、直接电导检测法;3、紫外吸收光度法;4、柱后衍生光度法;5、电化学法;6、与元素选择性检测器联用法。
五、离子色谱的应用
1、无机阴离子的检测
无机阴离子是发展最早,也是目前最成熟的离子色谱检测方法,包括水相样品中的氟、氯、溴等卤素阴离子、硫酸根、硫代硫酸根、氰根等阴离子,可广泛应用于饮用水水质检测、啤酒、饮料等食品的安全、废水排放达标检测、冶金工艺水样、石油工业样品等工业制品的质量控制。特别由于卤素离子在电子工业中的残留受到越来越严格的限制,因此离子色谱被广泛的应用到无卤素分析等重要工艺控制部门。
无机阴离子交换柱通常采用带有季胺功能团的交联树脂或其他具有类似性质的物质,常见的阴离子交换柱如Metrosep A supp 4-150, A supp 5-250等。常用的淋洗液为Na2CO3和NaHCO3按一定比例配置成的稀溶液,改变淋洗液的组成比例和浓度,可控制不同阴离子的保留时间和出峰顺序。
2、无机阳离子的检测
无机阳离子的检测和阴离子检测的原理类似,所不同的是采用了磺酸基阳离子交换柱,如Metrosep C1, C2-150等,常用的淋洗液系统如酒石酸/二甲基吡啶酸系统,可有效分析水相样品中的Li,Na,NH4+,K,Ca,Mg等离子。
3、有机阴离子和阳离子分析
随着离子色谱技术的发展,新的分析设备和分离手段不断出现,逐渐发展到分析生物样品中的某些复杂的离子,目前较成熟的应用包括:
1)生物胺的检测
Metrosep C1分离柱;2.5mM 硝酸/10%丙酮淋洗液; 3 µL进样,可有效分析腐胺、组胺、尸胺等成分,已经成为刑事侦查系统和法医学的重要检测手段。
2)有机酸的检测
Metrosep Organic Acids分离柱,MSM抑制器 ;0.5 mM H2SO4作为淋洗液,可有效分析包括乳酸、甲酸、乙酸、丙酸、丁酸、异丁酸、戊酸、异戊酸、苹果酸、柠檬酸等各种有机酸成分,在微生物发酵工业、食品工业都是简便有效的分离方法。
3)糖类分析
目前已经开发出各种糖类的分析手段,包括葡萄糖、乳糖、木糖、阿拉伯糖、蔗糖等多种糖类分析方法。在食品工业中的应用尤其广泛。
扩展阅读:
1.1、《离子色谱应用》2003,Claudia Eith,Maximilian Kolb
2.2、离子&电化学分析智库 Metrohm,2008
-
色谱字典(术语大全)
红杏 发布于 2008-02-13 23:15:01
亲和色谱法 affinity chromatography
离子交换色谱法 ion exchange chromatography,IEC
离子色谱法 ion chromatography
离子抑制色谱法 ion suppression chromatography
离子对色谱法 ion pair chromatography
疏水作用色谱法 hydrophobic interaction chromatography
制备液相色谱法 preparative liquid chromatography
平面色谱法 planar chromatography
纸色谱法 paper chromatography
薄层色谱法 thin layer chromatography,TLC
高效薄层色谱法 high performance thin layer chromatography,HPTLC
浸渍薄层色谱法 impregnated thin layer chromatography
凝胶薄层色谱法 gel thin layer chromatography
离子交换薄层色谱法 ion exchange thin layer chromatography
制备薄层色谱法 preparative thin layer chromatography
薄层棒色谱法 thin layer rod chromatography
液相色谱仪 liquid chromatograph
制备液相色谱仪 preparative liquid chromatograph
凝胶渗透色谱仪 gel permeation chromatograph
涂布器 spreader
点样器 sample applicator
色谱柱 chromatographic column
棒状色谱柱 monolith column monolith column
微粒柱 microparticle column
填充毛细管柱 packed capillary column
空心柱 open tubular column
微径柱 microbore column
混合柱 mixed column
组合柱 coupled column
预柱 precolumn
保护柱 guard column
预饱和柱 presaturation column
浓缩柱 concentrating column
抑制柱 suppression column
薄层板 thin layer plate
浓缩区薄层板 concentrating thin layer plate
荧光薄层板 fluorescence thin layer plate
反相薄层板 reversed phase thin layer plate
梯度薄层板 gradient thin layer plate
烧结板 sintered plate
展开室 development chamber
往复泵 reciprocating pump
注射泵 syringe pump
气动泵 pneumatic pump
蠕动泵 peristaltic pump
检测器 detector
微分检测器 differential detector
积分检测器 integral detector
总体性能检测器 bulk property detector
溶质性能检测器 solute property detector
(示差)折光率检测器 [differential] refractive index detector
荧光检测器 fluorescence detector
紫外可见光检测器 ultraviolet visible detector
电化学检测器 electrochemical detector
蒸发(激光)光散射检测器 [laser] light scattering detector
光密度计 densitometer
薄层扫描仪 thin layer scanner
柱后反应器 post-column reactor
体积标记器 volume marker
记录器 recorder
积分仪 integrator
馏分收集器 fraction collector
工作站 work station
固定相 stationary phase
固定液 stationary liquid
载体 support
柱填充剂 column packing
化学键合相填充剂 chemically bonded phase packing
薄壳型填充剂 pellicular packing
多孔型填充剂 porous packing
吸附剂 adsorbent
离子交换剂 ion exchanger
基体 matrix
载板 support plate
粘合剂 binder
流动相 mobile phase
洗脱(淋洗)剂 eluant,eluent
展开剂 developer
等水容剂 isohydric solvent
改性剂 modifier
显色剂 color [developing] agent
死时间 t0,dead time
保留时间 tR,retention time
调整保留时间 t'R,adjusted retention time
死体积 V0,dead volume
保留体积 vR,retention volume
调整保留体积 v'R,adjusted retention volume
柱外体积 Vext,extra-column volune
粒间体积 V0,interstitial volume
(多孔填充剂的)孔体积 VP,pore volume of porous packing
液相总体积 Vtol,total liquid volume
洗脱体积 ve,elution volume
流体力学体积 vh,hydrodynamic volume
相对保留值 ri.s,relative retention value
分离因子 α,separation factor
流动相迁移距离 dm,mobile phase migration distance
流动相前沿 mobile phase front
溶质迁移距离 ds,solute migration distance
比移值 Rf,Rf value
高比移值 hRf,high Rf value
相对比移值 Ri.s,relative Rf value
保留常数值 Rm,Rm value
板效能 plate efficiency
折合板高 hr,reduced plate height
分离度 R,resolution
液相载荷量 liquid phase loading
离子交换容量 ion exchange capacity
负载容量 loading capacity
渗透极限 permeability limit
排除极限 Vh,max,exclusion limit
拖尾因子 T,tailing factor
柱外效应 extra-column effect
管壁效应 wall effect
间隔臂效应 spacer arm effect
边缘效应 edge effect
斑点定位法 localization of spot
放射自显影法 autoradiography
原位定量 in situ quantitation
生物自显影法 bioautography
归一法 normalization method
内标法 internal standard method
外标法 external standard method
叠加法 addition method
普适校准(曲线、函数) calibration function or curve [function]
谱带扩展(加宽) band broadening
(分离作用的)校准函数或校准曲线 universal calibration function or curve [of separation]
加宽校正 broadening correction
加宽校正因子 broadening correction factor
溶剂强度参数 ε0,solvent strength parameter
洗脱序列 eluotropic series
洗脱(淋洗) elution
等度洗脱 gradient elution
梯度洗脱 gradient elution
(再)循环洗脱 recycling elution
线性溶剂强度洗脱 linear solvent strength gradient
程序溶剂 programmed solvent
程序压力 programmed pressure
程序流速 programmed flow
展开 development
上行展开 ascending development
下行展开 descending development
双向展开 two dimensional development
环形展开 circular development
离心展开 centrifugal development
向心展开 centripetal development
径向展开 radial development
多次展开 multiple development
分步展开 stepwise development
连续展开 continuous development
梯度展开 gradient development
匀浆填充 slurry packing
停流进样 stop-flow injection
阀进样 valve injection
柱上富集 on-column enrichment
流出液 eluate
柱上检测 on-column detection
柱寿命 column life
柱流失 column bleeding
显谱 visualization
活化 activation
反冲 back flushing
脱气 degassing
沟流 channeling
过载 overloading
-
ICP仪器的应用前景
Neo 发布于 2009-02-01 18:10:35
ICP–AES(包括ICP–MS)自发明以来,得到广大检测人员的喜爱,有关的文章以数十万篇计,涉及金属材料(包括贵金属、稀有金属)、非金属材料、矿石、土壤、核燃料、煤、石油及其产品、化肥、化工原料、半导体晶片、陶瓷材料、食品、药品、血液、水(纯水、废水)、空气等几乎所有材料中杂质(或粒子)的测定;但是,作为标准的ICP–AES(包括ICP–MS)测定方法却非常地稀少,国际标准才17个,其中钢铁的测定标准8个,其余为测定水、石油、空气等物质中杂质的方法;国家标准26个,其中22个为稀土的方法,其余为锌、刚玉、蜂蜜、润滑油的方法,行业标准有16个,其中检验检疫的标准有4个(SN):涉及钢铁、铝锭、锌精矿、金属猛,其它为:检定规程3个、HS、SH各3个,YS2个、SY一个,涉及金属材料和石油产品;其它国家的标准有:日本10个,俄罗斯3个,英国14个,法国13个德国15个,涉及的领域有:水、金属、石油产品、食品等。
ICP–AES(包括ICP–MS)测定方法由于步骤简单、一次性测定全部待测元素,是一个花费少,污染少,流程短的环保性方法,有着广宽的发展前景。元素分析是化学分析的一个重要组成部分,传统的元素分析方法包括分光光度法、原子吸收法(火焰与石墨炉)、原子荧光光谱法、ICP发射光谱法等。这些方法都各有其优点,但也有其局限性,例如:或是样品前处理复杂,需萃取、浓缩富集或抑制干扰;或是不能进行多组分或多元素同时测定,耗时费力;或是仪器的检测限或灵敏度达不到指标要求等。电感耦合等离子体质谱—ICP-MS(Inductively Coupled Plasma Mass Spectrometry)技术是几乎克服了传统方法的大多数缺点,并在此基础上发展起来的更加完善的元素分析法,因而被称为当代分析技术的重大发展。
ICP-MS的工作原理及其分析特性:在ICP-MS中,ICP作为质谱的高温离子源(7000K),样品在通道中进行蒸发、解离、原子化、电离等过程。离子通过样品锥接口和离子传输系统进入高真空的MS部分,MS部分为四极快速扫描质谱仪,通过高速顺序扫描分离测定所有离子,扫描元素质量数范围从6到260,并通过高速双通道分离后的离子进行检测,浓度线性动态范围达9个数量级从ppq到1000ppm直接测定。因此,与传统无机分析技术相比,ICP-MS技术提供了最低的检出限、最宽的动态线性范围、干扰最少、分析精密度高、分析速度快、可进行多元素同时测定以及可提供精确的同位素信息等分析特性。
ICP-MS的谱线简单,检测模式灵活多样:
(1)通过谱线的质荷之比进行定性分析;
(2)通过谱线全扫描测定所有元素的大致浓度范围,即半定量分析,不需要标准溶液,多数元素测定误差小于20%;
(3)用标准溶液校正而进行定量分析,这是在日常分析工作中应用最为广泛的功能;
(4)同位素比测定是ICP-MS的一个重要功能,可用于地质学、生物学及中医药学研究上的追踪来源的研究及同位素示踪。
ICP-MS发展简史及应用范围:电感耦合等离子体质谱ICP-MS,是20世纪80年代发展起来的新的分析测试技术。它以独特的接口技术将ICP-MS的高温(7000K)电离特性与四极杆质谱计的灵敏快速扫描的优点相结合而形成一种新型的元素和同位素分析技术,可分析几乎地球上所有元素。ICP-MS技术的分析能力不仅可以取代传统的无机分析技术如电感耦合等离子体光谱技术、石墨炉原子吸收进行定性、半定量、定量分析及同位素比值的准确测量等。还可以与其他技术如HPLC、HPCE、GC联用进行元素的形态、分布特性等的分析。随着这项技术的迅速发展,现已被广泛地应用于环境、半导体、医学、生物、冶金、石油、核材料分析等领域。
1. ICP-MS在环境样品分析中的应用
保护环境,实现可持续发展,正成为全世界的共识。世界各国政府及组织纷纷通过各种环境保护法规,对环境分析化学提出了越来越高的要求,环境分析化学样品多种多样,包括大气、水、岩石、砂土、泥土、污泥以及和生态环境相关的各种植物样品。世界各国的法规对这一些样品的浓度范围均作了严格的规定。为了保证所测定的结果的准确性,对于这些环境样品的分析所采用的分析仪器,分析方法、采样方法等也作了严格的法规规定,其中最典型的就是美国国家环保局所规定的ICP-MS技术用语,饮用水、地表水、地下水各种元素的EPA method 200.8和用于废水、固体废弃物、沉积物、泥土等样品中的各种元素分析的EPA method 6020。 随着环境法规对一些有毒有害元素的检测限的要求提高,对分析技术也提出了越来越多的需求。比如,根据建设部《城市供水业2000年技术进步发展规划》,新增水质指标项,其中要求检测的金属和非金属元素共有23种(新增12种):Fe、Mn、Cu、Zn、As、Se、Hg、Cd、Cr、(Ⅳ)、Pb、Ag、Al、Na、Ca、Mg、Si、Ba、B、Be、Ni、Sb、V、Co。这些元素的浓度范围大到数十甚至数百ppm(如Na、Ca、Mg、Si等),小至ppt级(如Hg)。由于检测项目大量增加,而且它们的基准和测限(浓度)都非常低,传统的分析方法如ICP-AES技术对Se、Hg、Be、As、Pb、Tl、U等元素不能达到检测限要求,必须与石墨炉原子吸收(GF-AAS)和汞冷原子吸收(CV-AAS)技术结合使用才能达到大部分元素的分析要求。而ICP-MS技术的出现,在某种程度上可以取代ICP-AES、GF-AAS和CV-AAS等分析,且可以测定它们均不能分析的饮用水标准中特殊要求的U和Tl。同时ICP-MS技术还可以直接测定海水中与环境污染或水文变化相关的多种元素。
2、ICP-MS与其他技术的联用及其在生命科学研究中的应用随着生命科学研究发展的需要,对环境卫生规划的新要求也不断提高,要求对元素分析的检测限也越来越低,对元素存在的形态要求也越明确。因为元素的形态不同,其作用的机理完全不同。因此,如果仅研究体系中元素的总含量,已经不足于研究该元素在体系中的生理和毒理作用。如Cr(Ⅲ)对人体大有益处,而Cr(Ⅵ)则会引起皮肤病、肺癌等,ICP-MS技术与离子色谱技术联用分别测定Cr(Ⅲ) 和Cr(Ⅵ)已经是十分成熟的方法,其检测限可以达ppt级,每个样品的操作时间不超过7分钟,操作简便,大大节省人力、物力。 HG-ICP-MS(氢化物发生器与ICP-MS)联用技术应用于海水中超痕量污染物如As、Se、Sb等易受干扰难测元素的分析具有优越性。 GC-ICP-MS技术已被用于多种污染物的形态分析,如船用涂料中有机Sn的影响,使牡蛎大量死亡,用GC-ICP-MS技术可分离出不同形态的有机锡代谢产物,从而推动了船用涂料的改进。在低泥中也曾用GC-ICP-MS联用技术分离测定二甲基铅、二乙基铅多种有机铅形态,推动汽车污染的环境迁移研究。生物对Hg的甲基化及富集作用是GC-ICP-MS技术的另一个应用范围。
高效毛细管电泳(CE)技术是目前最强有力的分离技术,CE与ICP-MS的强检测能力结合起来是将来联用技术最有潜力的应用领域。许多科学家都已在这一领域作了探索工作,并在生物化学领域有了一些具体的应用。 与环境化学、毒理学等生命科学研究关系最密切的应用当属高效液相色谱分离(HPLC)与ICP-MS联用技术。HPLC是一种具有高效的分离技术,尤其适用于热稳定性差、分子量大、极性较强的物质的分离。把HPLC与具有极低的检测限、宽的动态线性范围、干扰少、分析精密度高、速度快和可测定多元素等优点的ICP-MS联用,可用于研究中草药、藻类、鱼类、人类等生物体内含Cd、Se、As、Cu、Zn、Pb等元素与多种氨基酸、多肽和蛋白质结合的机理以及某些元素对酶的位点的作用过程。另外,某些维生素大环化合物和DNA片断与金属元素的作用也在HPLC-ICP-MS的联用技术发展中得到应用。能在复杂的基体中准确地分析微量、痕量元素同位素,同时将ICP-MS用作HPLC的检测器跟踪被测元素同位素在各形态中的信号变化,使得色谱图变得简单,有助于元素形态的确认及进行定量析。
3、展望
我国加入WTO后,新一轮的市场竞争将对国有产品提出更新的挑战。无论是产品的质量、生产、加工、包装到商标等,都必须与国际法规接轨,否则我们的产品将面临被淘汰的危险。为了适应新一轮的市场竞争的需求,让我们的产品走向国际市场,产品的质量控制在理论上必须提供和世界检测水平相符的可靠数据。不管是农业、医药、环保、食品、还是工业产品等,用ICP-MS进行这些产品中多元素的分析测定,可称之为是目前国际上在这一领域检测水平最高的分析技术,可为产品提供可靠的,国际技术领域认可的实验数据。因此,ICP-MS在未来的经济发展和科学研究中将发挥更为积极而重要的作用。
ICP-OES
ICP-AES法是以等离子体原子发射光谱仪为手段的分析方法,由于其具有检出限低、准确度高、线性范围宽且多种元素同时测定等优点,因此,与其它分析技术如原子吸收光谱、X-射线荧光光谱等方法相比,显示了较强的竞争力。在国外,ICP-AES法已迅速发展为一种极为普遍、适用范围广的常规分析方法,并已广泛应用于各行业,进行多种样品、70多种元素的测定,目前也已在我国高端分析测试领域广泛应用。
1 等离子体原子发射光谱仪的性能特点
1.1分析精度高
电感耦合等离子体原子发射光谱仪可准确分析含量达到10-9级的元素,而且很多常见元素的检出限达到零点几μg/L,分析精度非常高。对高低含量的元素要求同时测定,尤其对低含量元素要求精度高的项目,使用ICP-AES法非常方便。
1.2样品范围广
电感耦合等离子体原子发射光谱仪可以对固态、液态及气态样品直接进行分析,但由于固态样品存在不稳定、需要特殊的附件且有局限性,气态样品一般与质谱、氢化物发生装置联用效果较好,因此应用最广泛也优先采用的是溶液雾化法(即液态进样)。从实践来看,溶液雾化法通常能取得很好的稳定性和准确性。而在测试工作中,运用一定的专业知识和经验,采取各种化学预处理手段,通常都能将不同状态的样品转化为液体状态,采用溶液雾化法完成测定。溶液雾化法可以进行70多种元素的测定,并且可在不改变分析条件的情况下,同时进行多元素的测定,或有顺序地进行主量、微量及痕量浓度的元素测定。
1.3动态线性范围宽
一般的精密分析仪器都有它的线性范围(一般在103以下),以明确该类仪器准确测定的浓度区间(不同类型的仪器或同类不同生产厂家的仪器还有区别),如果待测元素的浓度过高或过低,就必须进行化学处理,如稀释或浓缩富集,使待测浓度位于误差允许的线性范围之内。因此,当常量元素和微量元素需要同时测定时,就增加了分析的难度,加大了工作量,而测定结果往往还不理想。
电感耦合等离子体原子发射光谱仪的动态线性范围大于106,也就是说,在一次测定中,既可测百分含量级的元素浓度,也可同时测10-9级浓度的元素,这样就避免了高浓度元素要稀释、微量元素要富集的操作,既提高了反应速度,又减少了繁琐的处理过程不可避免产生的误差。以粉煤灰为例,固态的粉煤灰经过适当的预处理(根据待测元素种类确定预处理方法)转化成液态,一次进样既可测定常量的铁、铝、钙等元素,也可同时测定微量的钒、钼等综合利用及环境评定时的影响元素,方便准确。
-
ICP-MS、ICP-AES 及AAS的比较
isabel 发布于 2009-01-30 22:21:50
诱人的ICP-AES的流行使很多的分析家在问购买一台ICP-AES是否是明智之举,还是留在原来可信赖的AAS上。现在一个新技术ICP-MS已呈现在世上,虽然价格较高,但ICP-MS具有ICP-AES的优点及比石墨炉原子吸收(GFAAS)更低的检出限。
这篇文章简要地论述这三种技术,并指出如何根据你的分析任务来判断其适用性的主要标准。
对于拥有ICP-AES技术背景的人来讲,ICP-MS是一个以质谱仪作为检测器的等离子体(ICP),而质谱学家则认为ICP-MS是一个以ICP为源的质谱仪。事实上,ICP-AES和ICP-MS的进样部分及等离子体是及其相似的。ICP-AES测量的是光学光谱(165~800nm),ICP-MS测量的是离子质谱,提供在3~250 amu范围内每一个原子质量单位(amu)的信息,因此,ICP-MS除了元素含量测定外,还可测量同位素。
检出限ICP-MS的检出限给人极深刻的印象,其溶液的检出限大部份为ppt级(必需记牢,实际的检出限不可能优于你实验室的清洁条件),石墨炉AAS的检出限为亚ppb级, ICP-AES大部份元素的检出限为1~10ppb,一些元素在洁净的试样中也可得到令人注目的亚ppb级的检出限。必须指出,ICP-MS的ppt级检出限是针对溶液中溶解物质很少的单纯溶液而言的,若涉及固体中浓度的检出限,由于ICP-MS的耐盐量较差,ICP-MS检出限的优点会变差多达50倍,一些普通的轻元素(如S、 Ca、 Fe 、K、 Se)在ICP-MS中有严重的干扰,也将恶化其检出限。
干扰
以上三种技术呈现了不同类型及复杂的干扰问题,为此,我们对每个技术分别予以讨论。
ICP-MS的干扰
1.质谱干扰
ICP-MS中质谱的干扰(同量异位素干扰)是预知的,而且其数量少于300个,分辩率为0.8amu的质谱仪不能将它们分辩开,例如58Ni 对58Fe、 40Ar对40Ca、 40Ar16O对56Fe或40Ar-Ar对80Se的干扰(质谱叠加)。元素校正方程式(与ICP-AES中干扰谱线校正相同的原理)可用来进行校正,选择性地选用一些低自然丰度的同位素、采用“冷等离子体炬焰屏蔽技术”或“碰撞池技术”可有效地降低干扰影响。
2.基体酸干扰
必须指出,HCl 、HClO4、H3PO4和H2SO4将引起相当大的质谱干扰。Cl+ 、P+ 、S+离子将与其他基体元素Ar+ 、O+ 、H+结合生成多原子,例如35Cl40Ar对75As 、35Cl16O对51V的叠加干扰。因此在ICP-MS的许多分析中避免使用HCl 、HClO4、H3PO4和H2SO4是至关重要的,但这是不可能的。克服这个问题的方法有:“碰撞池技术”、在试样导入ICP之前使用色谱(微栓)分离、电热蒸发(ETV)技术等,另外一个比较昂贵的选择是使用高分辨率的扇形磁场的ICP-MS,它具有分辨小于0.01amu的能力,可以清除许多质谱的干扰。
ICP-MS分析用的试液通常用硝酸来配制。3.双电荷离子干扰
双电荷离子产生的质谱干扰是单电荷离子M/Z的一半,例如138Ba2+对69Ga+,或208Pb2+对104Ru+。这类干扰是比较少的,而且可以在进行分析前将系统最佳化而有效地消除。
4.基体效应
试液与标准溶液粘度的差别将改变各个溶液产生气溶胶的效率,采用基体匹配法或内标法可有效地消除。
5.电离干扰
电离干扰是由于试样中含有高浓度的第I族和第II族元素而产生的,采用基体匹配、稀释试样、标准加入法、同位素稀释法、萃取或用色谱分离等措施来解决是有效的。
6.空间电荷效应
空间电荷效应主要发生在截取锥的后面,在此处的净电荷密度明显的偏离了零。高的离子密度导致离子束中的离子之间的相互作用,形成重离子存在时首先损失掉轻离子,例如Pb+对Li3+。基体匹配或仔细在被测物质的质量范围内选用内标有助于补尝这个影响,但这在实际应用是有困难的。同位素稀释法虽有效,但费用高,简单而最有效的方法是稀释样品。
ICP-AES干扰1.光谱干扰
ICP-AES的光谱干扰其数量很大而较难解决,有记录的ICP-AES的光谱谱线有50000多条,而且基体能引起相当多的问题。因此,对某些样品例如钢铁、化工产品及岩石的分析必须使用高分辩率的光谱仪。广泛应用于固定通道ICP-AES中的干扰元素校正能得到有限度的成功。ICP-AES中的背景较高,需离线背景校正,应用动态背景校正对增进准确度是很有效的。各种分子粒子(如OH)的谱峰或谱带对某些低含量的被测元素会引起一些分析问题,影响其在实际样品中检出限。
在ICP-MS中的背景是相当低的,典型的是小于5 C/S(计数/秒),这就是ICP-MS具有极好的检出限的一个主要理由。
2.基体效应
与ICP-MS一样,ICP-AES可以应用内标来解决例如雾化室效应、试样与标准溶液之间粘度差异所带来的基体效应。
3.电离干扰
仔细选用每个元素的分析条件或加入电离缓衡剂(如过量的 I 族元素)可以减少易电离元素的影响。
GFAAS干扰1.光谱干扰
使用氘灯背景校正的GFAAS有少许光谱干扰,但使用Zeeman 背景校正的GFAAS能去除这些干扰。
2.背景干扰
在原子化过程中,针对不同的基体,应仔细设定灰化步聚的条件以减少背景信号。采用基体改进剂有助于增加可以容许的灰化温度。在很多GFAAS应用中,与氘灯扣背景相比,Zeeman扣背景可得到更好的准确度。
3.气相干扰
这是由于被测物质的原子蒸汽进入一个较冷的气体环境而形成的。现在采用等温石墨管设计和平台技术,试样被原子化后进入一个热的惰性气体环境,可有效减少这种干扰。
4.基体效应
基体效应是被测物质在石墨管上不同的残留而生成的,它取决于样品的种类,应用基体改性剂和热注射能十分有效地减少这些影响。
容易使用在日常工作中,从自动化来讲,ICP-AES是最成熟的,可由技术不熟练的人员来应用ICP-AES专家制定的方法进行工作。ICP-MS的操作直到现在仍较为复杂,自1993年以来,尽管在计算机控制和智能化软件方面有很大的进步,但在常规分析前仍需由技术人员进行精密调整,ICP-MS的方法研究也是很复杂及耗时的工作。GFAAS的常规工作虽然是比较容易的,但制定方法仍需要相当熟练的技术。
试样中的总固体溶解量TDS
在常规工作中,ICP-AES可分析10%TDS的溶液,甚至可以高至30%的盐溶液。在短时期内ICP-MS可分析0.5%的溶液,但大部分分析人员乐于采用最多0.2%TDS的溶液。当原始样品是固体时,与ICP-AES,GFAAS相比,ICP-MS需要更高倍数的稀释,其折算到原始固体样品中的检出限显示不出很大优势的现象也就不令人惊奇了。
线性动态范围LDR
ICP-MS具有超过105的LDR,各种方法可使其LDR开展至108,但不管如何,对ICP-MS来说:高基体浓度会导致许多问题,而这些问题的最好解决方案是稀释,正由于这个原因,ICP-MS应用的主要领域在痕量/超痕量分析。
GFAAS的LDR限制在102~103,如选用次灵敏线可进行高一些浓度的分析。
ICP-AES具有105以上的LDR且抗盐份能力强,可进行痕量及主量元素的测定,ICP-AES可测定的浓度高达百分含量,因此,ICP-AES外加ICP-MS,或GFAAS可以很好地满足实验室的需要。精密度
ICP-MS的短期精密度一般是1~3% RSD,这是应用多内标法在常规工作中得到的。长期(几个小时)精密度为小于5%RSD。使用同位素稀释法可以得到很好的准确度和精密度,但这个方法的费用对常规分析来讲是太贵了。
ICP-AES的短期精密度一般为0.3~2%RSD,几个小时的长期精密度小于3%RSD。
GFAAS的短期精密度为0.5~5%RSD,长期精密度的因素不在于时间而视石墨管的使用次数而定。
样品分析能力
ICP-MS有惊人的能力来分析大量测定痕量元素的样品,典型的分析时间为每个样品小于5分钟,在某些分析情况下只需2分钟。Consulting实验室认为ICP-MS的主要优点即是其分析能力。
ICP-AES的分析速度取决于是采用全谱直读型还是单道扫描型,每个样品所需的时间为2或6分钟,全谱直读型较快,一般为2分钟测定一个样品。
GFAAS的分析速度为每个样品中每个元素需3~4分钟,晚上可以自动工作,这样保证对样品的分析能力。
根据溶液的浓度举例如下,以参考:
- 每个样品测定1~3个元素,元素浓度为亚或低于ppb级,如果被测元素要求能满足的情况下,GFAAS是最合适的。
- 每个样品5~20个元素,含量为亚ppm至%,ICP-AES是最合适的。
- 每个样品需测4个以上的元素,在亚ppb及ppb含量,而且样品的量也相当大,ICP-MS是较合适的。
无人控制操作ICP-MS,ICP-AES,和GFAAS,由于现代化的自动化设计以及使用惰性气体的安全性,可以整夜无人看管工作。
为了高的分析生产,整夜开机工作是可取的。
运行的费用
ICP-MS开机工作的费用要高于ICP-AES,因为ICP-MS的一些部件有一定的使用寿命而且需要更换,这些部件包括了涡轮分子泵、取样锥和截取锥以及检测器。对于ICP-MS和ICP-AES来讲,雾化器与炬管的寿命是相同的。如果实验室选用了ICP-AES来取代ICP-MS,那么实验室最好能配备GFAAS。GFAAS应计算其石墨管的费用。在上述三种技术中Ar气的费用是一笔相当的预算,ICP技术Ar费用远高于GFAAS。
基本费用
这是很难于限定的一个项目,因为费用是根据自动化程度、附件与供应商而定的。大概的估计ICP-AES是GFAAS的两倍,而ICP-MS 是ICP-AES的两倍。必须注意到附件的配置将打乱费用的估计。
另外必须考虑到超痕量分析需要一个干净的实验室和超纯的化学试剂,这些的费用不便宜。
附件
由于是快速扫描测定方式,ICP-MS能对多元素模式中的瞬间信号进行测量,这就为大量附件打开了出路,电热蒸法、激光消蚀、辉光放电及火花消蚀可以免除样品的溶解过程。有些附件可以将样品中的基体物质进行分离或进行预富集,例如:氢化法、色谱(高压液相HPLC,离子色谱,微柱)等技术。
用色谱来分离的好处在ICP-MS中得到完全的实现,它适合用于环保,毒理学,药品及食品中低浓度的被测物质。
虽然ICP-AES也能采用上述的某些附件,但由于这些附件的价格及有限的好处,因此,很少看到它们在ICP-AES的常规分析中应用。
概要对购买哪一种仪器提出建议是困难的,但根据你现在及将来工作的需要,回答列表1中的问题,将有助于你作出决定。
必需记着没有一种技术能满足你所有的要求,这些技术是相互补充的,永远存在某一种技术稍优于另一种技术的地方。
表2是这三种技术简单比较,表3是检出限的比较。
参考文献
1. G.Tyler, AA or ICP-which do you choose? Chemistry in Australia, Vo l 59, No 4, pp 150-152 , April 1992.
2. A.R. Date and A.L. Gray, Applications of ICP-MS, Blackie, Glasgow, UK, 1989.
3. K.E. Jarvis, A.L. Gray, and R.S. Houk, Handbook of ICP-MS, Blackie, Glasgow, UK, 1992.
4. M. Thompson, J,N. Walsh, Handbook of Inductively Coupled Plasma Spectroscopy, Blackie, Glasgow, UK, 1983.
5. J.E. Cantle, Atomic Absorption Spectroscopy, Elsevier, 1982.
6. Analytical Methods for Liberty ICP Spectrometer, Varian publication 85 100938 00, Chapter 5, pp 81-82.
7. J.01lesik, Elemental Analysis Using ICP-OES and ICP-MS, Anal. Chem. Vol 63 No 1, Jan 1 1991 PP 12A-21A.表1 对分析要求的核对表
1.每星期有多少样品需分析?
2.样品是那一种类别的(钢铁、岩石、地面水、土壤等)?
3.应用那种分解方法?
4.多少元素、那些元素需要测定?
5.元素的浓度范围?
6.可采用的试液的体积是多少?
7.要考虑那些选购件及附件?
8.同位素的测定对你是否重要?
9.有多少购买资金或每月的租金?
10.为了满足分析的需要日常开机的运行及基本费用有多少?
11.你有怎么样技术水平的工作人员?表2 ICP-MS, ICP-AES, GFAAS的简单比较
ICP-MS ICP-AES Flame AAS GFAAS 检出限 绝大部分元素非常杰出 绝大部分元素很好 部分元素较好 部分元素非常杰出 样品分析能力 每个样品的所有元素 2-6分钟 每分钟每个样品的5-30个元素 每个样品每个元素15秒 每个样品每个元素 4分钟 线性动态范围 108 105 103 102 精密度短期长期(4小时) 干扰光(质)谱化学(基体)电离质量效应同位素 固体溶解量(最大可容忍量) 0.1-0.4% 2-25% 0.5-3% >20% 可测元素数 > 75 >73 > 68 >50 样品用量 少 多 很多 很少 半定量分析 能 能 不能 不能 同位素分析 能 不能 不能 不能 日常操作 容易 容易 容易 容易 方法试验开发 需要专业技术 需专业技术 容易 需专业技术 无人控制操作 能 能 不能 能 易燃气体 无 无 有 无 操作费用 高 高 低 中等 基本费用 很高 高 低 中等/高 表3 检出限比较表(µg/L)
Element ICP-MS ICP-AES Flame AAS GFAAS As <0.050 <10 <500 <1 Al <0.010 <4 <50 <0.5 Ba <0.005 <0.2 <50 <1.5 Be <0.050 <0.2 <5 <0.05 Bi <0.005 <10 <100 <1 Cd <0.010 <1 <5 <0.03 Ce <0.005 <15 <200000 ND Co <0.005 <2 <10 <0.5 Cr <0.005 <3 <10 <0.15 Cu <0.010 <2 <5 <0.5 Gd <0.005 <5 <4000 ND Ho <0.005 <2 <80 ND In <0.010 <10 <80 <0.5 La <0.005 <1 <4000 ND Li <0.020 <1 <5 <0.5 Mn <0.005 <0.5 <5 <0.06 Ni <0.005 <2 <20 <0.5 Pb <0.005 <10 <20 <0.5 Se <0.10 <10 <1000 <1.0 Tl <0.010 <10 <40 <1.5 U <0.010 <20 <100000 ND Y <0.005 <0.5 <500 ND Zn <0.02 <0.5 <2 <0.01 ICP-MS, ICP-AES及火焰AAS的检出限的定义为:空白的三倍标准偏差。
GFAAS:灵敏度(0.0044吸光度) 以20ul样品进行测量。
-
原子吸收光谱分析中的干扰及消除
renzhihai 发布于 2008-01-09 14:50:37
虽然原子吸收分析中的干扰比较少,并且容易克服,但在许多情况下是不容忽视的。为了得到正确的分析结果,了解干扰的来源和消除是非常重要的。
1 物理干扰及其消除方法
物理干扰是指试样左转移,蒸发和原子化过程中,由于试样任何物理性质的变化而引起的原子吸收信号强度变化的效应。物理干扰属非选择性干扰。
1.1物理干扰产生的原因
在火焰原子吸收中,试样溶液的性质发生任何变化,都直接或间接的影响原子阶级效率。如试样的粘度生生变化时,则影响吸喷速率进而影响雾量和雾化交率。毛细管的内径和长度以及空气的流量同样影响吸喷速率。试样的表面张力和粘度的变化,将影响雾滴的细度、脱溶剂效率和蒸发效率,最终影响到原子化效率。当试样中存在大量的基体元素时,它们在火焰中蒸发解离时,不仅要消耗大量的热量,而且在蒸发过程中,有可能包裹待测元素,延缓待测元素的蒸发、影响原子化效率。物理干扰一般都是负干扰,最终影响火焰分析体积中原子的密度。
1.2消除物理干扰的方法
为消除物理干扰,保证分析的准确度,一般采用以下方法:
a 配制与待测试液基体相一致的标准溶液,这是最常用的方法。
b 当配制与待测试液基体相一致的标准溶液有困难时,需采用标准加入法。
c 当被测元素在试液中浓度较高时,可以用稀释溶液的方法来降低或消除物理干扰。
2.光谱干扰及其消除方法
原子吸收光谱分析中的光谱干扰较原子发射光谱要少得多。理想的原子吸收,应该是在所选用的光谱通带内仅有光源的一条共振发射线和波长与之对应的一条吸收线。当光谱通带内多于一条吸收线或光谱通带内存在光源发躬垢非吸收线时,灵敏度降低且工作曲线线性范围变窄。当被测试液中含有吸收线相重叠的两种元素时,无论测哪一种都将产生干扰。
a光谱通带内多于一条吸收线
如果在光谱内存在光源的几条发射线,而且被测元素对这几种辐射光均产生吸收,这就产生干扰。也就是所谓的多重谱线干扰,以过渡元素较多。若多重吸收线和主吸收线波长差不是很小时,通过减小狭缝来克服多重谱线的干扰。但波长差很多小时,通过减小狭缝仍难消除干扰,并且可能使信噪比大大降低,此时需别选谱线。
b光谱通带内存在光源发射的非吸收线
待测元素的非吸收线出现在光谱通带内,这非吸收线可以是待测元素的谱线,也可能是其它元素的谱线.此时产生的干扰使灵敏度降低和工作曲线弯曲。造成这种干扰的原因有几种:
1具有复杂光谱的元素本身就发射出单色器难以分开的谱线;
2多元素空芯阴极灯因发射线较复杂而存在非吸收干扰;
3光源阴极材料中的杂质所引起的非吸收干扰;
4光源填充的惰性气体的辐射线引起的非吸收干扰。克服这种干扰常用方法是减小狭缝宽度,使光谱通带小到步以分离掉非吸收线,但使信噪比变坏。这时可以改用其它分析线,虽灵敏度较低,但允许较大的光谱通带,有利于提高信噪比。
3 吸收线重叠干扰
火焰中有两种以上原子的吸收线与光源发射的分析线相重叠时产生邻近线干扰,这种干扰使结果偏高。当分析元素的吸收线和共存元素的吸收线完全重叠,而分析元素的含量很低时,测得的只是共存元素的吸收信号。当分析元素的分析线中心位置和共存元素的吸收线的中心位置稍有偏离,但仍有相当程度的重叠,此时得于的吸收信号仍有很大一部分是共存元素产生的。当共存元素的吸收线和分析元素的吸收线稍有重叠时,吸收信号中仍有小部分是共存元素产生的。只有分析元素的吸收线和共存元素的吸收线完全分离时,共存元素才不产生干扰。Co253.649对Hg253.652r的干扰是典型的吸收线重叠干扰。
理论研究和实验结果表明,干扰的大小取决于吸收线重叠程度,干扰元素的浓度及其灵敏度。当两种元素的吸收线的波长差小于0.03nm时,则认为吸收线重叠干扰是严重的。若重叠的吸收线是灵敏线,即使相差0.1nm,干扰也会明显表现出来。当然这种干扰还和干扰元素的浓度及单色仪的分辨率有关。有一些谱线,在理论上是重叠线,但实验中并没有观察到干扰。有可能是干扰元素在测定条件下原子化效率低而未能产生足够的基态原子,也可能这些干扰元素的吸收线灵敏度很低,所以在通常情况下表现不出来。消除这种干扰一般是选用其它的分析线或预分离干扰元素。

