







伴随诊断介绍(二)
(四)《指导原则》的核心内容
在这份长达48页的《体外伴随诊断设备与治疗产品的共同开发指导原则》(草案)中,最重要的部分莫过于第三大部分——共同开发过程的原则,其中包括体外诊断和治疗产品的监管条例 、潜在合作发展项目的IVD验证计划、治疗产品临床试验设计注意事项、晚期期治疗产品的IVD开发注意事项、标签注意事项以及上市前注意事项等。
其中,关于治疗产品临床试验与IVD的产品验证试验中,需要注意的事项尤为重要,本文仅用以下两个图表进行解读。在进行伴随诊断产品开发及申报过程中,可以参照大纲内容进行文档索引。
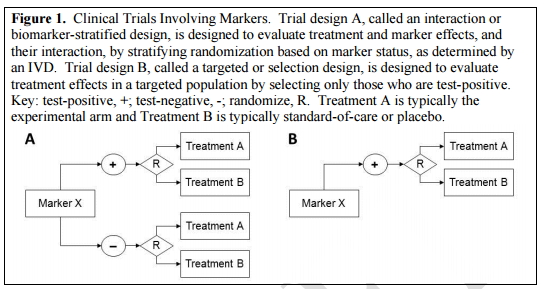
注:图中“+”指阳性,“-”指阴性,R指随机,A代表实验组,B代表安慰剂组
众所周知,在开发伴随诊断产品时,离不开对标记物进行临床试验的分析与验证。
图1A叫做“生物标志物的分层设计”或“生物标志物的相互作用”,用以评价治疗产品与标记物的效果以及它们之间的相互作用,该方法作为一种IVD产品测试,是基于标志物随机状态下的分层试验;
图1B叫做“靶向设计”或“选择设计”,旨在评估特定人群中,谁能够对治疗方法产生阳性效果的测试。
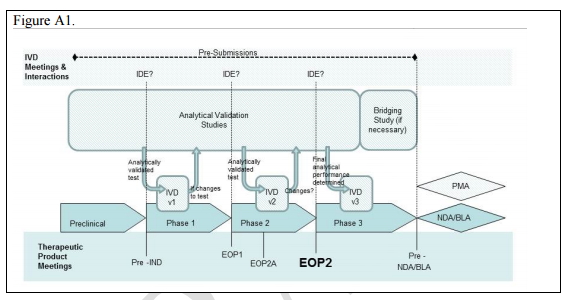
共同开发过程的关键点
毋庸置疑,一种治疗方法的伴随诊断的高效合作方式必须是建立在:两个产品研发项目团队的积极协作,甚至包括FDA系统所有相关部门间的合作与相互交流。
如图1所示:在某个药物/生物制品从临床前研究到NDA/BLA(新药上市许可/生物制品许可申请)过程中,先后需要经过临床前研究,1、2、3期临床试验。
在这个过程中,相应的体外诊断测试也需要同步进行(见图中的IVD v1、IVD v2、IVD v3)。其中针对在临床前研究的完成时(Pre-IND)、1期临床试验结束点(EOP1)和2期临床试验结束点(EOP2)所开发的伴随诊断测试都需要进行医疗器械临床试验申报(IDE)。
在上述的过程中,如何设计相应控制点的目标?如何衡量出现的偏差?谁应对哪些失误负责?哪些信息反馈价值最大、最经济实用等等。具体说来,企业在设计试验时就需要要制定一些客观的标准。
(五)FDA已经批准的28款伴随诊断产品
根据生物探索的前期统计(截止到2016年5月24日),FDA已经批准了一些伴随诊断检测。这些检测由罗氏、雅培、QIAGEN、DAKO等公司推出,主要利用qPCR、原位杂交、免疫组化等方法筛查一些肿瘤相关突变,以协助医生选择适当的疗法。
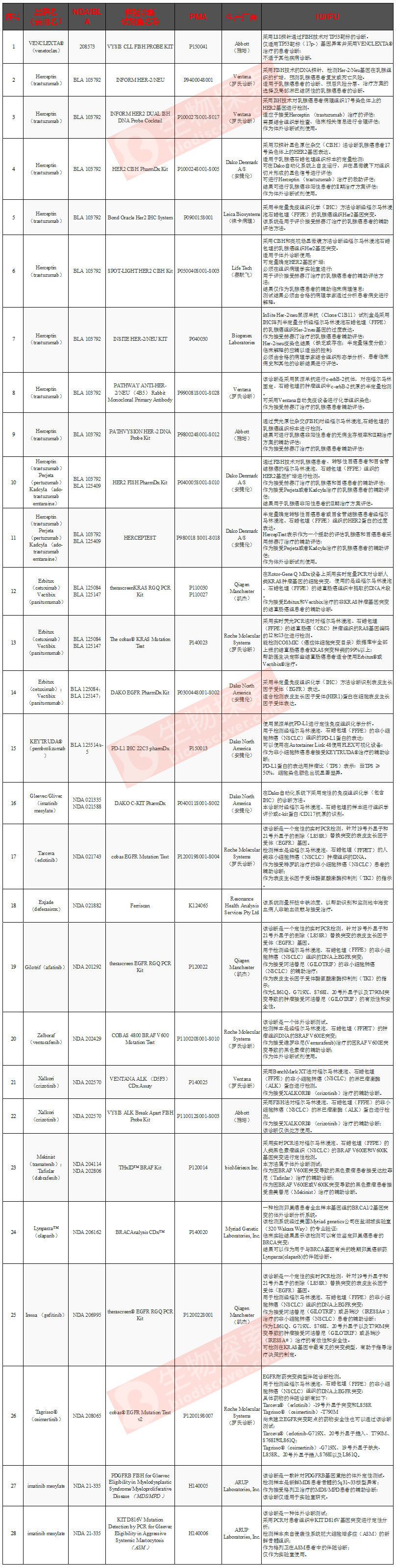
备注:NDA指新药申请;BLA指生物制剂许可申请;PMA指上市前申请许可;IU指预期用途;IFU指使用适应症。
(六)展望:伴随诊断,体外诊断市场发展最快的领域
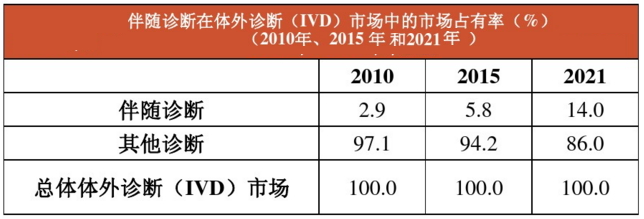
由上图可见,伴随诊断是一个新兴市场,预计到2021年,伴随诊断将占整个体外诊断(IVD)市场14%的份额。
目前已经有越来越多的医疗器械(IVD)公司开始与制药企业展开合作,为后者的在研药物/生物制品开发伴随诊断试剂盒。
作为精准医疗的重要分支,伴随诊断是对于预测患者针对特定药物的治疗反应至关重要。
对尚未或正在准备开展伴随诊断合作,拓宽新兴市场的药企&诊断公司而言,仔细研究FDA发布的这份长达48页的“葵花宝典”,必将让“研发——试验——上市”过程更加顺利。
-
仪器推荐

-
仪器推荐

-
仪器推荐

-
仪器推荐

-
仪器推荐

-
企业风采

-
企业风采

-
产品技术

-
产品技术

-
会议会展

-
产品技术

-
焦点事件

















