Western blot 标准操作规程(SOP)
关键词:western blot 聚丙烯酰胺 分离胶 积层胶 电泳
目的:蛋白质的检测与分析
【原理】
一个基因表达终极结果是产生相应的蛋白质(或酶)。因此检测蛋白质是测定基因表达的主要标志,检测蛋白质的方法很多,除ELISA法外,也可用与检测DNA和RNA相类似的吸印方法。前两法有“南”和“北”之意,故本法遂被延伸称为Western(西)印迹法,该法能用SDS聚丙烯酰胺凝胶电泳分辨出与专一抗血清结合的专一性蛋白质。将聚丙烯酰胺凝胶上分辨出的蛋白质转移到硝酸纤维素膜上并与第一抗体共孵。第一抗体专一地与待分离蛋自质的抗原决定簇结合,然后用另一种蛋自质,如135I-蛋白A或辣根过氧化物酶连接的山羊抗IgG检测已结合上去的抗体。本法所需时间6小时或过夜。
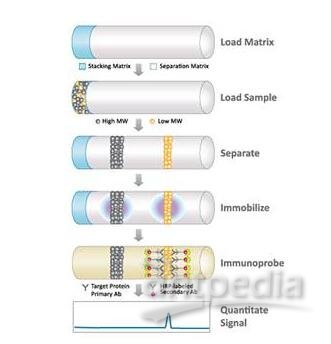
蛋白质的聚丙烯酰胺凝胶电泳
几乎所有蛋白质电泳分析都在聚丙烯酰胺凝胶上进行,而所用条件总要确保蛋白质解离成单个多肽亚基并尽可能减少其相互间的聚集。最常用的方法是将强阴离子去污剂SDS与某一还原剂并用,并通过加热使蛋白质解离后再加样于电泳凝胶上。变性的多肽与SDS结合并因此而带负电荷,由于多肽结合SDS的量几乎总是与多肽的分子量成正比而与其序列无关,因此SDS多肽复合物在聚丙烯酰凝胶电泳中的迁移只与多肽的大小相关。在达到饱和的状态下,每克多肽约可结合1.4克去污剂,借助已知分子量的标准参照物,则可测算出多肽链的分子量。
SDS聚丙烯酰胺凝胶电泳大多在不连续缓冲系统中进行,其电泳槽缓冲液的pH值与离子强度不同于配胶缓冲液,当两电极间接通电流后,凝胶中形成移动界面,并带动加入凝胶的样品中所含的SDS多肽复合物向前推进。样品通过高度多孔性的积层胶后,复合物在分离胶表面聚集成一条很薄的区带(或称积层)。曲于不连续缓冲系统具有把样品中的复合物全部浓缩于极小体积的能力,故大大提高了SDS聚丙烯酰胺凝胶的分辨率。
最广泛使用的不连续缓冲系统最早是由Ornsstein(1964)和Davis(1964)设计的,样品和积层胶中含Tris-Cl(pH6.8),上下槽缓冲液含Tris-甘氨酸(pH8.3),分离胶中含Tris-Cl(pH8.8)的。系统中所有组分都含有0.1%的SDS(Laemmli, 1970),样品和积层胶中的氯离干形成移动界面的先导边界而甘氨酸分子则组成尾随边界,在移动界面的两边界之间是一电导较低而电位滴度较陡的区域,它推动样品中的多肽前移并在分离胶前沿积聚,此处pH值较高,有利于甘氨酸的离子化,所形成的甘氨酸离子穿过堆集的多肽并紧随氯离子之后,沿分离胶泳动。从移动界面中解脱后,SDS多肽复合物成一电位和pH值均匀的区带泳动穿过分离胶,并被筛分而依各自的大小得到分离。
【常用试剂】
1)30%丙烯酰胺/0.8% N,N’-亚甲丙烯酰胺
将30克丙烯酰胺和0.8克N,N’-亚甲丙烯酰胺溶于总体积为60ml的水中,加热至37℃溶解之,补加水至终体积为100ml。0.45µm微孔滤膜过滤除菌,查证该溶液pH应不大于7.0,置棕色瓶中保存。
小心:丙烯酰胺具有很强的神经毒性并可通过皮肤吸收,其作用具有累积性。称量丙烯酰胺和N,N’-亚甲丙烯酰胺时应戴手套和面具。可认为聚丙烯酰胺无毒,但也应谨慎操作,因为它还可能含有少量未聚合材料。
2)4×Tris•Cl/SDS, pH8.8,
在300mlH2O中溶解91g Tris碱(1.5mol/L),用1mol/L调节pH至8.8, 补加H2O至体积500ml。用0.45um滤膜过滤溶液,再加入2g SDS[0.4%(w/v)],于4℃可保存1月。
3)4×Tris•Cl/SDS, pH6.8,
在40mlH2O中溶解6.05g Tris碱(0.5mol/L),用1mol/L调节pH至6.8, 补加H2O至体积100ml。用0.45um滤膜过滤溶液,再加入0.4g SDS[0.4%(w/v)],于4℃可保存1月。
4)4×SDS电泳缓冲液
Tris base 24.2 g
Glycerin 115.3 g
20%SDS 20 ml
加水至总体积1000ml。
应用时稀释4倍即为1×SDS电泳缓冲液(Tris 0.05M, Glycerin 0.38M, SDS 0.1%)。
5)TEMED (N,N,N’,N’-四甲基乙二胺)
TEMED通过催化过硫酸铵形成自由基而加速丙烯酰胺和N,N’-亚甲丙烯酰胺的聚合。
6)10%过硫酸铵
过硫酸铵提供驱动丙烯酰胺和亚甲丙烯酰胺聚合所必需的自由基。可用去离子水配制小量10%(w/v)的贮存液并保存于4℃。由于过硫酸铵会缓慢分解,故应隔周新鲜配制。
【步骤】
1)按厂商的使用指南用两块干净的玻璃,平板和0.75mm垫片组装电泳装置中的玻璃平板夹层,并固定在灌胶支架上。
2)按表7.1配制分离胶液体并脱气,然后加入10%的过硫酸铵和TEMED,轻轻搅拌混匀。
表7.1 聚丙烯酰胺分离胶的配制
试剂成分 配制不同浓度分离胶所需试剂(ml)
5% 8% 10% 12% 15%
Acry:Bis (30:0.8) 2.50 4.00 5.00 6.00 7.50
4×Tris•Cl/SDS, pH8.8 3.75 3.75 3.75 3.75 3.75
H2O 8.75 7.25 6.25 5.25 3.75
10%过硫酸铵 0.05 0.05 0.05 0.05 0.05
TEMED 0.01 0.01 0.01 0.01 0.01
按所需分离的蛋白质分子大小选择合适的丙烯酰胺百分比浓度,一般地,5%的凝胶可用于60~200kDa的SDS变性蛋白质分子的分离,10%用于16~70kDa,15%用于12~45kDa。
3)用一根巴斯德吸管立即将分离胶液体沿夹层中一条垫片的边缘加入于玻璃平板夹层中,至凝胶约5cm高为止。样品体积少于10μl不需灌制积层胶。
4)用另一根已斯德吸管,先从一边的垫片,再从另一边垫片往夹层的液面顶部缓缓加入一层水饱和异丁醇(厚约1cm)。让凝胶在室温聚合30min。
聚合后,可见在顶层异丁醇与凝胶的界面间有一清晰的折光线,胶的聚合失败往往问题在于过硫酸按或TEMED,或两者都有。
5)倾去顶层的异丁醇,并以1×Tris-Cl/SDS, pH8.8缓冲液冲洗凝胶的顶部表面,尽量用吸水纸吸干。
6)按表7.2配制积层胶液体,用吸管将液体沿一条垫片加入到玻璃平板夹层,直至夹层的顶部。
表7.2 聚丙烯酰胺积层胶的配制
GEL%(3.9%) Total (10.05ml)
Acry:Bis(30:0.8) 1.30 ml
4×Tris•Cl/SDS, pH6.8 2.50 ml
H2O 6.10 ml
10%过硫酸铵 0.05 ml
TEMED 0.01 ml
7)将0.75mm厚的梳子插入夹层的积层胶液体中,必要时,再补加积层胶液体充盈剩余空间。让积层胶室温聚合30min。
8)在具螺口盖的微量离心管中,用2×SDS加样缓冲液按1:1(v/v)稀释待测蛋白质样品,于100℃煮沸5-10min。如样品是蛋白质沉淀物,加入50~100µl 1×SDS加样缓冲液溶解之,并同样在100℃煮沸5-10min。按供应商的使用指南用2×SDS加样缓冲液溶解蛋白质分子量标准混合物。
对于0.3cm宽的加样孔,推荐加样体积以不超过20µl 为宜。用考马斯亮蓝染色法显迹,成分很复杂的蛋白质混合物需加25~50µg,而样品中只有一种或不多的几种蛋白的话,只需1~10µg蛋白量。采用银染显迹时,样品用量可减小10~100倍(按样品的复杂程度在小于20µl的体积溶有0.01~0.5ng蛋白样品不等)。
2×SDS加样缓冲液(loading buffer)配制:
成 分 体积 (ml)
0.5M Tris•Cl (pH6.8) 12.5
20%SDS 11.5
Glycerin 10
2% Blue-Bromo-phenol 2.5
β-mercaptoethanol * 5.0
加H2O至总体积50 ml,分装
*β-mercaptoethanol 在临用前加入。
9)小心拔出梳子,避免撕裂聚丙烯酰胺凝胶加样孔。取出梳子后,以1×SDS电泳电泳缓冲液冲洗加祥孔,并以此缓冲液充满之。
10)按厂商指南将凝胶板固定到电泳装置的上缓冲液室(上槽),同时往下缓冲液室(下槽)加入推荐量的1×SDS电泳缓冲液。
11)将固定于上槽的凝胶板放入下槽中,并往上槽加入部分电泳缓冲液至刚好淹没凝胶的加样孔。
12)用带平嘴针头的50µl注射器将同样浓度的蛋白质样品等体积加入到样品孔中,小心加样使样品在孔的底部成一薄层,对照孔加入蛋白质分子量标准样品,如有空置的加样孔,须加等体积的空白1×SDS样品缓冲液,以防相邻泳道样品的扩散。
13)再往上槽加入余下的1×SDS电泳缓冲液。此操作缓慢小心,以防冲起样品孔中的样品。
14)连接电源,对于0.75mm厚的垂直板电泳,先在60V下电泳至溴酚蓝染料从积层胶进入分离胶,再将电压调至120V继续电泳至溴酚蓝到达凝胶底部为止。
15)关闭电源并撤去连接的导线,弃去电泳缓冲液。连同上槽一起将凝胶夹层取出。
16)将凝胶定位以便识别加样的顺序,将凝胶板从上槽解离出来,放在一叠吸水纸或纸巾上。
17)小心将封边的垫片抽出一半,并以此为杠杆橇起上面的玻璃平板,使凝胶暴露出来。
18)小心从下面的玻璃平板上移出凝胶,在凝胶的一角切去一小块以便在染色及干胶后仍能认出加样次序,接着就可进行蛋白质的检测。
19)转膜
将凝胶面与负极相连,硝酸纤维素膜与正极相连。(100V,1小时)要放于4℃。
1. For transfer of proteins smaller than 20 kDa, transfer proteins from gel to PVDF (polyvinylidene difluoride) membrane at 1Amp constant current for 45 mins or equivalent (250mAmp for 3 hours or 500mAmp for 90 minutes) in transfer buffer (25mM Tris, 190mM glycine, 20% MeOH).
2. For transfer of proteins smaller than 120 kDa, transfer proteins from gel to PVDF membrane at 1Amp constant current for 1 hour or equivalent (250mAmp for 4 hours or 500mAmp for 2 hours) in transfer buffer (25mM Tris, 190mM glycine, 20% MeOH).
3. For proteins larger than 120kDa, transfer to PVDF membrane at 1Amp constant current for 90 minutes or equivalent (250mAmp for 6 hours or 500mAmp for 3 hours) in transfer buffer + SDS (25mM Tris, 190mM glycine, 20% MeOH, 0.05% SDS).
4. For Proteins larger than 250kDa, transfer to PVDF membrane at 1Amp constant current for 1 hour and 45 minutes or equivalent (500mAmp for 3.5 hours) in transfer buffer + SDS (25mM Tris, 190mM glycine, 20% MeOH, 0.05% SDS).
Add transfer (or CAPS) buffer to the transfer unit according to manufacturer1s directions. Transfer over night with a setting of 20V / 40 mA or for 3 hours at 70V/160 mA. The transfer process needs to take place under cold temperatures to prevent the gel from sticking to the membrane. This can be accomplished either by using a water cooling core in the transfer unit or placing the entire unit at 4?. Please follow the manufacturer1s recommendations
20)、清洗
将硝酸纤维素膜放入1X TBST中清洗3次,每次5分钟。摇床摇动。(这步忘了!)
21)、封闭
1. Remove the blot from the transfer apparatus or staining tray and immediately place into blocking buffer (1% BSA, 10mM Tris pH 7.5, 100mM NaCl, 0.1% Tween 20).
2. Incubate the blot for 1 hour at 37℃, 2 hours at room temperature, or overnight at 4℃.
22)、一抗孵育
一抗用TBST1:1000稀释,将硝酸纤维素膜放入其中,37℃孵育1.5小时,(或4℃过夜)摇床摇动。
Probe with primary antibody in TTBS/1% NFDM for 1 hr. at room temperature. Primary antibody should be diluted as specified on product data sheets. If these values are not available, use the following guidelines for initial experiments: apply antisera or ascites at 1:500 to 1:5,000, apply purified primary antibodies at a concentration of 1 ug/ml.
23)、清洗
将硝酸纤维素膜放入1X TBST中清洗3次,每次5分钟。
24)、二抗孵育
二抗用TBST1:8000稀释,将硝酸纤维素膜放入其中,37℃孵育1小时,摇床摇动。
25)、清洗
同22,之后,用1X TBS在清洗2次,每次5分钟,以洗去膜上的Tween 20,因为它可以阻碍底物的沉积。
26)、显色
按如下配方配制显色液:
1mL水+1滴(大约50微升)试剂A(之后混匀)+1滴试剂B+1滴试剂C
将硝酸纤维素膜放入其中孵育,一旦显色,立即放入去离子水中终止反应。
Considerations:
Perhaps the most common problem in western blotting is the occurrence of background staining. The best remedy for high background (in many cases) is simply to dilute the primary antibody further. Other solutions include ensuring that detergent is used in the blocking reagent, using an alternate blocking reagent (casamino acids, BSA, serum), and decreasing the amount of protein applied to the electrophoresis gel.
• Occurrences of extra bands in the blot can be resolved by several strategies. Run a control blot omitting the primary antibody to determine if the secondary antibody is the source of the problem. Replacing the secondary antibody with a different lot or a similar reagent from a different source can provide resolution. Spurious bands below the targeted molecular weight suggest that the protein is being degraded in the experiment; inclusion of protease inhibitors can help.
• No or low signal can be remedied by loading more protein in the gel or increasing the amount of primary and/or secondary antibody applied to the blot.
• Some antibodies will not bind in the presence of detergent; consult the datasheet for each antibody prior to performing any procedure