肝损伤与肝细胞凋亡
【关键词】 肝细胞
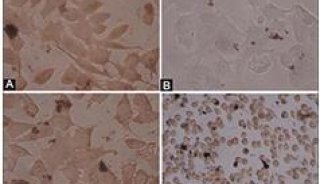
肝细胞凋亡的病理学特征为肝细胞的嗜酸性变,又称嗜酸性坏死。表现为个别肝细胞胞浆的嗜伊红性增强,胞核固缩直至消失,形成嗜酸小体(eosinophilic body),脱离肝细胞索坠入肝窦或窦间隙。周围可出现CTL、枯否细胞,将凋亡小体吞噬、降解。嗜酸性小体主要出现在急性病毒性肝炎或慢性病毒性肝炎活动期,也可出现在药物性肝炎。关于细胞死亡过程的研究,近年来已成为国内外生物学、医学研究的一个热点 [1~3]。本文复习近年来有关肝损伤与肝细胞凋亡机制国内外文献作如下综述。
肝细胞死亡可分为凋亡和坏死,二者在形态、发生机制以及概念上都有很大的差别。由体内、体外因素触发的细胞内预存的死亡程序而导致细胞程序性死亡的过程称为细胞凋亡(apoptosis,APO)。又称程序性死亡(programmed cell death,PCD)。凋亡属于细胞自主的生理性死亡,主要表现为:细胞皱缩,细胞核染色质浓缩,胞核空泡状,染色质DNA断裂成片段,但细胞膜仍保持完整。凋亡常发生于生理环境。而坏死多由肝细胞受各种致病因素作用导致,表现为细胞肿胀,细胞膜丧失完整性,进而形成噬酸性小体或者溶解。肝脏中有许多刺激可启动肝细胞的死亡,如缺血、再灌等引发的缺氧、活性氧代谢物、药物等毒性化合物以及乙醇、药物、病毒性肝炎等引发的TNF释放和自身免疫型、药物诱导及病毒性肝炎等诱发的Fas-L,Granzyme B等等。一些刺激启动后需要死亡受体(death receptors)及细胞内信号机制的参与,从而引发程序性细胞死亡;而另一些刺激则可能绕过死亡受体参与的级联反应或者直接嵌入级联反应的下游起作用。
肝细胞凋亡的过程,细胞凋亡是一种级联反应。TNF-α或者Fas-L等配体可能引发凋亡,这些配体可与细胞表面的死亡受体结合,也可被细胞内的事件激活并插入级联反应。肝脏中细胞的凋亡发生在各种细胞类型中,包括肝实质细胞、Kupffers细胞、内皮细胞和星状细胞等。研究表明,多种类型的肝损伤,包括暴发性肝衰竭、病毒性肝炎、肝硬化、自身免疫性肝病以及肝脏肿瘤的发病机制都和凋亡密切相关[2],因此,研究肝细胞的凋亡对各类肝病的治疗有重要意义。
胱冬肽酶(caspase)参与的凋亡途径是细胞很重要的死亡途径。 经典的凋亡级联反应从死亡受体(death receptor)开始。死亡受体包含一个位于胞质内的死亡结构域(death domaine),死亡结构域可与适应蛋白(adaptor protein)结合形成骨架结构,并可使起始或效应胱冬肽酶自我活化。当Fas-L与Fas结合或TNF-α与TNF-R1结合后,单个的受体分子形成三聚体使死亡受体聚集成簇。然后Fas与Fas相关的死亡受体蛋白(Fas associating protein with death domain,FADD),TNF-R1与TNF受体相关的死亡受体蛋白(TNFR associating protein with death domain,TRADD)结合,然后与FADD结合。FADD与caspase 8结合,使其活化。TRADD也与受体相互作用蛋白(RIP)和TNF受体相关因子2(TRAF2)等蛋白质结合。这些结合可活化激酶,继而活化NF-κB等转录因子,从而激活相关基因的转录,使肝细胞对抗TNF-α引起的凋亡。
在凋亡的级联反应中,一类重要的物质就是胱冬肽酶(caspase)。这类蛋白水解酶的活性位点(active site)有还原型半胱氨酸(cys),水解位点位于底物的天冬氨酸(asp)残基后。caspase在细胞内以无活性的酶原形式存在,它们可被分为3组:(1)与细胞因子产生相关的ICE-like caspase (如caspase 1,4,5,13);(2)死亡信号或者启动caspase,它传送死亡程序,而并无不可逆水解蛋白质的功能(如caspase2,8,9,10);(3)死亡效应或者执行caspase,它断裂所选择的底物,这样,当细胞本身的修复系统不能发挥作用时,它就可以断裂细胞骨架或细胞核。在不同的细胞类型中,凋亡有两条不同的信号传导途径[3]:(1)启动caspase被激活后,产生一强的信号反应,然后直接活化效应caspase;(2)当启动caspase活化的程度不够充分时,它需要一个放大过程,这一过程通常有线粒体的参与。当起始caspase作用于Bcl-2家族中某些成员(如Bid)或者神经酰胺使之被修饰,然后该分子(Bid或者神经酰胺)在线粒体分子上定位,改变线粒体外膜通透性,使细胞色素C和其他与凋亡相关的线粒体蛋白的前体如凋亡诱导因子(AIF)等由线粒体内释放出来。细胞色素C与胞质骨架蛋白(如apaf-1)和procaspase9结合,这一复合物一般被称作凋亡小体(apoptosome)。在ATP及其水解酶存在时,细胞色素C-apaf-1复合物寡聚化,使得procaspase9(信号caspase)自身活化,然后caspase9激活caspase3(效应caspase)。这样促进了细胞凋亡效应相互的作用。
线粒体与肝脏损伤:近年来关于线粒体的研究表明,线粒体在细胞死亡中起着关键的作用。线粒体功能障碍已成为肝脏损伤的一个重要机制,它在细胞凋亡以及坏死过程都起着重要的作用[4]。细胞色素C是线粒体释放的前凋亡(proapoptotic)因素。在细胞质,细胞色素C与凋亡激活因子Ⅰ(apoptotic-activating factor-1)结合,再与ATP(或dATP)结合,激活caspase 9,然后激活caspase 3,caspase 3刺激凋亡的效应途径,导致ADP-核糖聚合酶(PARP)断裂,核小体间DNA水解,细胞皱缩,染色体边缘化,核小叶形成。caspase 也参与线粒体释放细胞色素C的上游过程。TNF-α和Fas-L与其受体的结合激活caspase8。Caspase 8断裂Bid,然后定位至线粒体,诱导细胞色素C的释放。线粒体的瞬时通透性孔道(MPT)[5~7]是线粒体内膜一种可由铜离子诱导的可逆性通道,它在跨膜线粒体蛋白释放至胞液的过程中起到重要作用。MPT使得线粒体内外膜连接处一种复杂的蛋白质大通道开放,该孔道具有非特异性以及高传导性,它允许分子量<1500U的分子自由通过,以达到平衡。该孔道的传导性非常之大以至于当只有几个,甚至只有1个孔道开放时就足以使线粒体去极化,氧化磷酸化解耦联,线粒体严重肿胀,因此对细胞来说,MPT的开放可能导致致命的后果。在许多凋亡模型中,MPT与线粒体膜电位的严重破坏以及细胞色素C的释放同时发生,这提示这些事件有一定的关联。MPT不仅在程序性细胞死亡中起重要作用,而且也是坏死性死亡的关键步骤。因此,线粒体在多种细胞死亡形式中起到关键的连接作用。细胞若发生凋亡则需要未损伤的线粒体产生足够的ATP方能维持,而MPT的快速发生将影响多数的线粒体,最终使ATP耗竭,氧化应激大量发生。所有这些都将抑制细胞的凋亡,但对由于离子梯度及钙离子活化等功能的丧失造成的细胞肿胀以及溶解性坏死有促进作用。因此,广泛的MPT可使线粒体功能大量丧失,继而使细胞坏死。
与死亡程序相关的另一重要成分是Bcl-2 家族[8]。这一家族的不同成员对凋亡或坏死可能具有促进或抑制两种不同的作用,其主要作用靶点是线粒体。Bcl-2 家族成员或存在于细胞之中,或以非活性态与膜结合。其中主要的抗凋亡成分都是膜结合态的,有一些可与线粒体膜结合。它们的重要功能之一就是调节MPT。Bcl-2家族成员能形成同源或异源二聚体,不同的二聚体可能对凋亡产生不同的作用(诱导或抑制)[9]。除了形成二聚体外,某些成分还具有离子通道的作用,这对促进或抑制线粒体的肿胀具有重要作用。这一家族之间的相互作用及其复杂性提示尚有很多方面值得我们对其去研究。
与肝细胞凋亡相关的基因有数十种,分为凋亡抑制性基因、促凋亡基因和双向调控基因。三者处于动态平衡状态,共同控制着细胞的增殖或凋亡。凋亡抑制性基因主要为Bcl-2,其抗凋亡机制主要为抑制线粒体释放促凋亡因子、维持钙稳定、抑制caspase活化、直接抗氧化及抑制促凋亡蛋白Bax、Bak的细胞毒作用。促凋亡基因的代表为P53基因,主要在细胞周期的G1期发挥作用,如发现DNA有缺陷,则阻止细胞进入细胞周期,同时启动DNA修复机制,如DNA损伤不可修复时,则启动凋亡机制,将损伤细胞清除。当P53发生突变后,则失去促凋亡的功能。P53基因是促使肿瘤发生的癌基因之一。凋亡的双向调控基因c-myc是一种癌基因,即能诱导细胞增殖,也能诱导细胞凋亡。
【参考文献】
1 Garcia Tsao G, Sanyal AJ, Grace ND, et al. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis.Hepatology,2007,46(3):922-938.
2 Runyon BA. A pill a day can improve survival in patients with advanced cirrhosis.Gastroenterology,2007,133(3):1029-1031.
3 Salerno F, Gerbes A, Gines P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis.Gut,2007,56(9):1310-1318.
4 Pessayre D,Mansouri A,Haouzi D, et al. Hepatotoxicity due to mitochondrial dysfunction. Cell Biol Toricol,1999,15:367-373.
5 Lemasters JJ,Nieminen AL,Qian T,et al.The mitochondrial permeability transition in cell death:a common mechanisn in ncerosis and apoptosis.Biochim Biophys Acta,1998,1366:177-196.
6 Lemasters JJ.Mechanisums of hepatic toxicity V.Necrapoptosis and the mitochondrial permeability transition:Shared pathways to necrosis and apoptosis. Am J Physiol,1999,276:1-6.
7 Susin SA,Zamzami N,Kroemer G.Mitochondria as regulators of apoptosis:Doubt no more. Biochim Biophys Acta,1998,1336:151-165.
8 Adams JM,Cory S.The Bcl-2 protein family:Arbiters of cell survival.Science,1998,281:1322-1326.
9 Desagher S,Osen Sand A,Nichols A,et al.Bid induced conformational change of Bax is responsible for mitochondrial cytochrome corelease during apoptosis.J Cell Biol,1999,144:891-901.