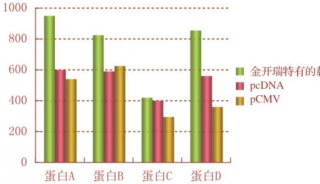
下述方法是对 Graham 与 Van der Eb(1973) 建立的磷酸转方法的改进,用高分子量基因组 DNA 代替了质粒 DNA。这一方法对建立稳定的携带补充宿主染色体基因突变的转染基因的细胞系尤其有用(Segeetal.1984,Kingsley et al.1986)。本实验来源于分子克隆实验指南(第三版)下册,作者:〔美〕J. 萨姆布鲁克 D.W. 拉塞尔。
| 实验材料 | 指数生长的哺乳动物细胞培养物 |
|---|
| 试剂、试剂盒 | CaCl2甘油HEPES 盐缓冲液异丙醇NaCl基因组 DNA带有选择性标志的质粒细胞生长培养基 |
|---|
| 仪器、耗材 | 聚乙烯试管Shepherd’s crook组织培养皿 |
|---|
| 实验步骤 | 材料
缓冲液与溶液
贮存液、缓冲液与试剂的成分见附录 1。
稀释贮存液至所浓度。
CaCl2(2mol/L)
过滤除菌,分装成 5 ml 的等份冻存。
甘油(15%V/V)1XHEPES 盐缓冲液。
HEPES 盐溶液过滤除菌,使用前加入离压灭绝的 15% 的甘柚。
HEPES 盐缓冲液
21 mmol/LHEPES
0.7 mmol/LNa2 HPO4
137 mmol/LNaCl
5 mmol/LKCl
6 mmol/L 右旋葡葡糖
调节 pH7.10, 过滤除菌;分装成 25~50 ml 的等份冻存。转染前溶解,取少量测定 pH, 如有必要重新调至 pH7.10。
异丙醇
NaCl(3mol/L)
过滤除菌, 室溫保存。
核酸与寡核苷酸
基因组 DNA
按第 6 章方案 3 所述,从适当的细胞中制备高分子量 DNA, 用 TE 缓冲液(pH7.6) 稀释 DNA 至 100ug/m。转染每 90 mm 培养皿的细胞大约需要 20~25ug DNA。
转染细胞前,基因组 DNA 必须切成 45~60kDa 大小(见步骤 2 与 3)。最好在预实验时按下述方法确定剪切 DNA 的条件:用规格为 22#的针头剪切 2 ml 的各等份高分子量 DNA, 剪切不同次数(如 3,4,5 或 6 次)。进行 0.6%(m/V) 琼脂糖凝胶电泳,溴化乙锭或 SYBR 金染色。用线性噬菌体λDNA 的单体或二聚体作标记,优化其他搡作步骤需要将剪切成适当大小的 DNA 用无细咆的培养皿进行第 9 步操作。
带有选择性标志的质粒
选用,见第 3 与 12 步的注释。
培养基
细胞生长培养基(完全培养基与选择性培样基)
特殊设备
聚乙烯试管(12 ml)
Shepherd’s crook(弯头吸管)
末端带勾的硅化玻璃巴氏吸管
组织培养皿(90 mm)
细胞与组织
指数生长的哺乳动物细胞培养物
方法
1. 实验第一天,以每 90 mm 培养皿 5x105 个细胞的密度,用适当的含血清生长培养基将指数生长的细胞(如 CHO 细胞)铺至培养皿上,在含 5%CO2 的 37°C 孵箱内培养细胞 16 h。
2. 第二天,按预实验确定的次数用规格为 22#的针头将高分子质量的 DNA 剪切成 45kb 至 60kb 大小的片段(见材料部分关于基因组 DNA 的说明)。
每 90 mm 培养血的细胞需 20~25ug 基因组 DNA 转染。
3. 加 0.1 体积的 3 ml/LNaCl 与 1 体积的异丙醇沉淀 DNA。用弯头吸管收集 DNA。将沉淀沿试管壁导入另一装有 HEPES 缓冲液(每 12~15ugDNA lml) 的试管内。37°C 轻轻旋转试管 2 h 以重新溶解 DNA。在继续下面的实验前确保所有的 DNA 都已溶解。
如果共转染选择性标志(见下面第 12 步的注释), 则加适当质粒溶液于基因组 DNA 中至终浓度 0.5ug/ml。
4. 取每等份 3 ml 的剪切的基因组 DNA 置于 12 ml 聚乙烯试管中(每等份 DNA 可用于转染 2 个培养皿)。
不同细胞系的转染效率不同,不同的筛选方法也会导致转染效率的差异。一般来说,要得到 3~10 个稳定转染体必须转染大约 15~20 个培养皿的 CHO细胞。
5. 轻轻旋转各等份基因组 DNA,滴加入 120ul 2mol/LCaCl2 以形成磷酸钙-DNA 沉淀物。室温下放置试管 15~20 min, 溶液将变混浊,但不会形成可见的沉淀团块。
6. 吸去两个培养皿中的培养基(步骤 1), 每一培养皿中轻轻加入 1.5 ml 磷酸钙-DNA 沉淀物,小心地旋转培养皿使沉淀覆盖细胞层,室温下放置 20 min, 期间旋转培养皿一次。
7. 轻轻加入 10 ml 预热(37°C) 的生长培养基于各培养皿。在含 5%CO2;的 37°C 孵箱内培养细胞 6 h。
8. 重复步骤 5~7, 直至所有培养皿的细胞都有磷酸钙-DNA 沉淀物覆盖。
9. 培养 6 h 后,在光学显微镜下观察各培养皿的细胞。可见"胡椒状"沉淀附于细胞上,沉淀不是粉末状的,也不是团块状的。
在光镜下要靠经验判断什么是"胡椒状"沉淀. 如果此时看到粉末状或团块状的沉淀,则终止实验。此步操作不能形成"胡椒状"沉淀或第 5 步溶液能变浑浊是由于所用 HEPES 缓冲盐溶液 pH 值不正确,第 5 步培养时间过长,或者是 CaCl2 或 DNA 浓度不合适造成的。
10. 大多数情况下,此时用甘油处理会增强转染效率。用甘油使细胞休克:
a. 吸去含磷酸钙-DNA 沉淀物的培养基。
b. 每培养皿细胞中加入预热至 37°C 的 1xHEPE 配置的 15% 甘油 3 ml。室温下培养不超过 3 min。
应注意 HEPE 缓冲液中的甘油不能与细胞接胜时间过长。最佳时间范围很窄,不同细胞系不同,不同实验室不同,因此一次只处理几个培养皿, 并应考虑到吸去甘油缓冲液的时间。不要超过最佳培养时间。时间要以秒来计算!
c. 吸去甘油 HEPE 缓冲液,快速用 10 ml 预热的培养基洗涤培养皿 2 次。
d. 加入 10 ml 预热的生长培养基,于含 5%CO2 的 37°C 孵箱内培养 12~15 h。
11. 用 10 ml 新配制的生长培养基换液。继续在 5%CO2 的 37°C 孵箱内过夜培养。
12. 此时(第 4 天)显微镜下观察细胞应呈正常形态。可用胰酶消化细胞并用选择性培养基重新培养,继续培养 2~3 周以形成互补与/或抗性克隆。每 2~3 天更换培养基一次。
筛选期的长短、重铺细胞的密度与筛选条件均取决于互补或筛选的突交或基因《此步重铺细胞的密度最好在每 90 mm 培养皿 2.5X105 至 1X106 细胞之间。此参数根据用不同数目的细胞铺培养皿(不作转染)并进行筛选的经验而确定。从逻辑上讲,应该使细胞密度尽可能大同时又可有效地杀死非互补或无抗性的细胞。
共转染(如,用赋予宿主 G418 抗性的质粒)可用于区分转染体与回复体。有些突变细胞系的回复频率可高达 (即百万分之一的细胞). 假阳性结果就很成问转染頻率通常为 2X10-7,共转染频率为,10-8。将选择标志(如 G418 抗性)与要筛选的突变/基因偶联可以消灭假阳性。详细说明见信息栏中的共转化。
13. 然后,克隆并繁殖单个克隆以用于检测(方法见 Jakby and PaStan l979 及第 16 章 16.18 步骤 7)。
用预冷的甲醇固定细胞 15 min, 然后室温下用 10% Giemsa 染色 15 min, 流水冲洗,这样可以纪录细胞克隆数目。Giemsa 染液应使用前用磷酸盐缓冲液或水新配置,用 Whatman1 号滤纸过滤。

 |
|---|