多羟基反应单克隆抗体的鉴定、生产和使用——免疫亲和层析
实验步骤
一、多羟基反应单克隆抗体
我们最先使用了一类特殊的单克隆抗体, 并将其用于免疫亲和层析。这些抗体可以用于温和的免疫亲和层析, 因为洗脱条件只需要联合使用无离液盐和低分子质量的多羟基化合物 (多元醇),此条件下蛋白质呈非变性状态。我们把这种抗体归类为多羟基反应单克隆抗体 (PR-mAb)。
我们的实验室研究蛋白质转录。因此,我们分离出的大部分 PRmAb,是原核或真核系统中与转录相关的
mAb。对于主攻分离的科学家,真核生物转录系统具有重大的挑战性, 因为实际上许多因子都是多亚基蛋白质。例如,真核生物的 RNA 聚合酶
n(RNAPn) 含 12 个亚基 (12 个不同基因产物)。然而,从启动子起始转录,rnapn 还需要转录因子
TFIIA、TFIIB、TFIID、TFIIE、TFIIF 和 TFIIH[综述见 Woychik 和 Hampsey(2002)]。除
TFIffi 外, 所有这些转录因子均是由两个或两个以上的亚基组成。大蛋白质复合物是使用 PR-mAb
免疫亲和层析的理想对象。我们已经研发用于大肠杆菌 RNAPCThompsonetal.,1992) 和真核生物
RNAPII(Thompsonetal.,1990) 的 PR-mAb,以及针对一些真核生物转移因子 (表 28.1) 的 PR-mAb。

PR-mAb 技术应用中最值得注意的是,采用我们的 PR-mAb8WG16 纯化的酵母 RNAPIKEdwardsetal.,1990) 已用于蛋白质复合物结晶(Crameretal.,2000)。其他实验室已成功分离出多种 PR-mAb, 并应用于纯化目标蛋白质 (Jiangetal.,1995;Lynchetal.,1996;Nagyetal.,2002)。也可以对已经确定的 mAb 进行筛选,将其用于多羟基反应。
目前已经发表了一些关于 PR-mAb 的综述(BurgessandThompson,2002;Thomp-sonandBurgess,1996,2 0 01;Thompsonetal.,2 00 6)。本章中,我们将对其中一些方法进行更新。我们还将介绍采用非常便宜的细胞培养系统制备大量 RP-mAb 的方法,以及以同样的方式用小鼠腹水制备极少量的 mAb。最后,根据抗体的多羟基反应特点,以重组 DNA 的方法, 用 RP-mAb 的表位标记无关目标蛋白质,可进而通过温和的多羟基-洗脱法纯化该蛋白质。
1.PR-mAb 特性
(1) 通过筛选大量单抗(218 个抗原特异孔),我们预计 PR-mAb 占单克隆抗体的 5%~10%(Thompsonetal.,1992)。
⑵在主孔 (master-well) 阶段进行筛选可以立即鉴定 PR-mAb。
(3)PR-mAb 不受小鼠 IgG 亚型的限制。也已在大鼠的 mAb 中鉴别筛选了 PR-mAb(R.R.Burgess, 数据未公开发表)。
(4) 多数 PR-mAb 受不同种盐和多羟基化合物组合的影响。最常见的是 0.75mol/L 硫酸铵或 0.75mol/L 氯化钠与 30%~40% 丙二醇联合使用。
(5)PR-mAb 可以是高亲和力抗体。事实上, 在稀释溶液中,高亲和力是保证 mAb 与抗原有效结合的首要条件。洗脱时成为低亲和力抗体。
(6) 多数 (不是全部)PR-mAb 受不同种盐和多羟基化合物组合的影响。经验证,盐包括硫酸铵、氯化钠、乙酸钠和谷氨酸钾! 多元醇包括丙二醇、乙二醇、2,3-丁二醇,有时甘油也可。
(7) 盐和多经基化合物通常用来作为蛋白质稳定剂, 其温和洗脱作用可以保留抗原的生物学特性及结构的完整性,甚至可用于多亚基复合物(CrameretaL,2000;Thomp.sonetal.,1990)。
2.mAb 来源
我们采用杂交瘤技术制备 mAb(Harl o wandLane,1988),即将抗原刺激的小鼠脾细胞和骨髓瘤细胞株融合。实验证明应首选高免小鼠; 仅 5%~10% 的单抗是 PR-mAb,因此需要大量的原始杂交瘤细胞才可能分离出 PR-mAb。但可以使用其他方法制备 mAb, 如反转录病毒感染浆细胞 (Largaespadaetal.,1”6),或通过重组技术构建抗体库。
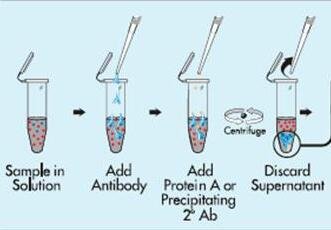
无论何种来源的抗体,必须具备抗原特异性。我们发现,标准 (ELISA) 是行之有效的方法。接着我们通过改良的 ELISA 法筛选抗体,即 ELISA-elution 检测法。该方法是在标准 ELISA 的基础上,在加入酶联二抗前,增加了用盐和多羟基化合物联合处理特异性的抗原抗体复合物的步骤。接着,用盐/多羟基化合物解离抗原抗体复合物后, 通过底物反应进行定量。ELISA-elution 检测法的一般程序如下所述。
3.PR-mAb 的 ELISA-elution 检测法
(1) 抗原包被聚苯乙烯微孔板。通常每孔加入 50 含有 30~100ng 抗原的磷酸盐缓冲液 (PBS,PH7.4)。室温孵育 Ih,保证足够的时间使抗原与聚苯乙烯结合。
(2) 每孔用 200 斗含 1% 脱脂奶粉的 PBSd%BLOTTO) 封闭。通常在 4°C 封闭过夜, 室温下 2 h 即可。
(3) 将待测抗体 (50(uL) 加入相邻的两个孔,室温孵育 Ih。通常细胞培养液中的抗体可以直接使用 D 但是,亲和力非常髙的抗体,或滴度非常高的抗体制剂应稀释至非饱和水平使用。
(4) 用含 0.l%tween20 的 PBS(PBST) 洗板 5 次,去除未结合的抗体。
(5) 对照孔,每孔加入 100fJLTE 缓冲液 [50 mmol/LTris-HCl(pH7.9),0.1 mmol/LEDTA]; 待测孔,每孔加入 100 含 0.75mol/L 硫酸铵和 40% 丙二醇的 TE 缓冲液。
EUSA 板室温孵育 20 min,偶尔 (约每 5 min) 轻拍 ELISA 板一侧以混合溶液。
(6) 用 PBST 洗板 5 次。
(7)1%BLOTTO 稀释商品化辣根过氧化物酶标记的二抗 (一般 1:2000),每孔加人 50|uL。室温孵育 Ih。
(8) 用 PBST 洗板 10 次。
(9) 向孔中加人适当底物。我们使用的是 100^含有 0.03%H2O2 和 0.4 mg/mL 邻苯二胺 (OPD) 的 0.05mol/L 柠檬酸缓冲液 (pH5.0)。
(10) 酶联板在室温下孵育 5~15 min。成对 (TE 和 TE+盐/多元醇对于每种单抗)终止反应,每孔加人 50fxLImol/LH2SO4。
(11) 酶联仪读取吸光值。对于()PD, 检测波长为 490nm。与仅加入 TE 缓冲液的对照孔相比 (图 28.1A),经多羟基化合物和盐处理的待测孔中,PR-mAb 的吸光值将降低约 50%。如果无酶联仪可用, 吸光值通常降低约 50% 时目测也是很明显的。
说明
(1)PR-mAb 筛选可以在主孔阶段 (master-wellstage) 进行操作。杂交瘤细胞筛选用于特异性抗体制备,随后立即进行 PR-tnAb 筛选。初步筛选可以在 100 细胞培养液中进行 (50 对照缓冲液和 50;^含多羟基化合物及盐的缓冲液)(Thompsonetal.,1992)。其中一项研究中, 在主孔阶段,仅一次融合我们就筛选了 200 个以上杂交瘤细胞用于制备 PR-mAb。
(2) 抗原结合于微孔板上可导致抗原结构的变化。这样可能会导致在溶液中埋藏于蛋白质内部的表位暴露, 进而导致假阳性,因为在溶液中 mAb 与目标不反应,这种 mAb 不适用于免疫亲和层析。
(3) 当有数毫升细胞培养液可用时, 可以在一块板上检测不同浓度的多元醇和盐对 mAb 的影响(图 28.1B)。

4.连续培养法生产 mAb
在许多情况下,已经不再鼓励用腹水生产小鼠 mAb 的做法。因此,我们已经研发了替代的方法用于生产免疫亲和层析所需的大量
mAb。这部分将介绍一种已经证实非常适合用于这种规模的抗体制备方法。该操作程序中, 使用一种商品化的由 IntegraBiosciencesAG
公司(瑞士)生产的 CELLineFlask350(CL350) 细胞培养室。美国 ArgosTechnologies
公司(Elgin,IL) 和 BioracoInternational 公司(Framingham,MA)
也提供这种产品。我们发现这个产品易于使用,能够制备 10~50 mg 抗体,且相同的杂交瘤细胞可重复使用数次。需要具备的条件包括:
标准的无菌细胞培养技术、细胞培养罩 (cellculturehood) 和 WC 恒湿二氧化碳培养箱 (5%)。这种培养瓶的示意图如 28.2A
所示。常规操作规程如下所述。

(1) 准备细胞培养基。准备两种略有差异的培养基用于两个 CL350 培养皿。杜尔伯科改良伊格尔培养基 [Dulbecco’smodifiedeaglemedium(DMEM)] 含有谷氨酰胺和高浓度的葡萄糖 (Gibco/Invitrogen#11965),再加入 Immol/L 丙酮酸納 (Sigma 公司)、100 单位/mL 青霉素、100fxg/mL 链霉素 (Gibco/Invitrogen),作为两种培养基的基础成分。在细胞生长室中,DEME 需添加上述成分和 15% 灭活的胎牛血清(Hycl0ne 公司)。
在营养维持室中,DMEM 需添加上述成分和 5% 胎牛血清的 DMEM 培养液。我们将细胞生长室用培养基称为「完全培养基」(completemedium),将营养维持室用培养基称为「营养培养基』』(nutrientmedium)。
(2) 准备用于接种培养瓶的接种液。从液氮罐中取出一管杂交瘤细胞,37°C 迅速解冻。将杂交瘤细胞按照约 2XIO4 个细胞/mL 密度用完全培养基铺板。我们使用含有 20 mL 培养基的 10 cm 细胞培养板。
(3)接种培养瓶。准备 5 mL 新鲜的完全培养基,含有 8XIO6~20XIO6 个处于对数生长期的活细胞。在细胞铺于细胞生长室前, 将 25 mL 营养培养基加至营养维持室 (绿盖)中,浸润营养维持室和细胞生长室之间的膜。悬浮细胞, 并用 10 mL 的血清移液管吸出。拧松绿盖,将吸管稳稳插入细胞生长室,向细胞生长室(白盖) 中接种 5 mL 悬浮液。
缓慢的上下吹打液体,在将液体吹回室内前使气泡上升,进而除去气泡。更换白盖并拧紧。向营养维持室中加人 35 0 mL 营养培养基,拧紧绿盖。
(4) 收获细胞生长室和培养维持。每隔 3~7 天更换营养维持室中的营养培养基。
拧松绿盖,将 10 mL 移液管插入细胞生长室, 上下吹打液体,彻底混合细胞。然后将整个细胞生长室中的液体全部转移至离心管。由于渗透通量,体积可能大于 5 mL。取样后用血细胞计数法进行细胞计数,台盼蓝进行活细胞染色检测。取出细胞生长室中的内容物离心沉淀细胞。取出合并含有 rnAb 的培养基, 将该上清液冻存以备日后纯化。用新鲜的完全培养基重悬细胞。可能需要根据初始接种密度、增长率和换液频率,将细胞分成几份 (通常为 1:2~1:4)。拧松绿盖,将 5 mL 的细胞倒回至培养皿中(白盖)。去除气泡和拧紧白盖。向营养室中加入 350 mL 培养基,彻底拧紧绿盖。
(5) 培养方式建立后,每隔 3~7 天收获 IntegraCL350 瓶一次。收获时间间隔取决于杂交瘤细胞增长速率和杂交瘤细胞对培养瓶环境的适应能力。这一特点具有细胞系依赖性。
说明
(1) 用 IntegraCL350 瓶培养杂交瘤细胞的效果很好, 每毫升细胞培养收获上清液中,可以得到 mAb0.5~Img。通常可连续培养 1 个月左右。
(2) 处理液体:将培养基在 37°C 水浴预热。这有助于避免培养瓶中的冷凝作用以及细胞的温度休克向细胞培养皿 (白盖) 添加或取出液体时, 要先拧松营养培养室的绿盖,以防止留有空气。放人培养箱孵育之前始终拧紧培养瓶的白盖和绿盖。建议使用 IOmL 血清移液管进行操作。将培养基吸出并加人新鲜的培养基, 为营养维持室换液。
(3) 接种的最低细胞浓度为 1.5X106 个细胞/mL[步骤 (2)]。我们尝试降低营养培养基中所需的胎牛血清,但未获成功。一些市场上的新型无血清培养基可以用做营养培养基。
(4) 监测细胞数量和状态很重要 [步骤 (3)]。有助于确定是否分离细胞,以减少细胞数量,避免总活细胞数大于 I.OXlO8 个。如果细胞活力大大降低,则需要增加换液频率。
(5) 在连续培养过程中,由于每次收获均分离细胞,引起细胞死亡而导致活细胞比例 [步骤 (4)] 下降。培养期末, 仅有 30%~40% 活细胞是非正常终止的。
(6)—些杂交瘤细胞在长期连续培养时是不稳定的, 并失去产生抗体的能力。因此,在培养期间应监测抗体生成。我们采用标准 ELISA 检测。
(7)CELLine 培养瓶的信息可以在 www.integrabiosciences,COM/celline 获得。
5.抗体纯化
为将抗体与载体上可用反应位点间的结合最大化, 需要纯化抗体,至少部分纯化也是有益的。可以用多种不同的方法纯化抗体。其中最经济实用的方法是分子排阻色谱或离子交换层析。常用的纯化方法是利用 ProteinA、ProteinG,或两者混合进行亲和层析, 但这种方法耗费较髙。小鼠 111 八 1) 属于免疫球蛋白化 04 个亚类之一:^0 1、:^02&、龟 02 匕和 IgG3。小鼠 IgG2a、IgG2b 和 IgG3 可以用 ProteinA 层析柱纯化。小鼠 IgGl 与 ProteinA 结合的效果差。与小鼠 IgG2a 不同,小鼠 IgGl 与 ProteinG 结合效果更好,而 IgG2b 与 ProteinA 和 ProteinG 均可结合。
根据我们的经验,大部分典型融合实例中的单抗都是 IgGl 亚类。这里我们介绍一种 DEAE 柱 (WhatmanDE52),可以用于纯化小鼠 IgGmAb,该层析柱既经济,又简单。
对于 mAbIgGl, 这种方法特别有效。事实上,在我们的操作中,每个纯化后的 igG1mAb,经检测纯度均可达 90% 左右。此外,许多小鼠的 IgG2a 和 IgG2b 抗体可以通过这种方法纯化。
(1) 无论是腹水或 CELLine 细胞培养上清液中的抗体均需采用硫酸铵沉淀。向 mAb 中加饱和硫酸铵溶液至奶% 饱和度。冰上搅拌浆体 2 0 min。通过这种方式沉淀 mAb,同时大部分血清白蛋白留在溶液中。
(2) 离心分离沉淀 (约 6000 尽,10 min)。向沉淀中加入抗体用缓冲液 [50 mmol/LTris-HCKpH6.9),25 mmol/LNaCl], 加入量为抗体初始体积的 l/4(CELLine 培养瓶制备抗体).1/2(腹水)。
(3)15 min 左右可溶解沉淀,离心使其澄清 (约 6000IOmin)。
(4)4°C 条件下,将步骤 (3) 得到的上清液用 IL 抗体用缓冲液透析过夜。
(5) 澄清的上清液用于 DE52 柱层析,按照下面「说明」中事项准备层析柱。层析柱用抗体用缓冲液 (pH6.9) 平衡, 该 pH 条件下,大部分 mAb 流穿,而多数杂质会与层析柱结合。10 mL 原料可用 5 mLDE52 柱。
(6) 收集原料中未与层析柱结合的组分 (约 ImL),每个组分样品进行 SD&PAGE 检测。CL350 培养瓶制备 mAb8RB13(IgGl 型单抗),纯化产物的 SDS^PAGE 结果如图 28.2B 所示。根据抗体纯度合并分离组分。
(7) 用 5 mL 含 0.5mol/LNaCl 的抗体用缓冲液洗脱层析柱,PAGE 分析前保存洗脱液。
说明
(1)DE52 的准备工作十分重要。虽然生产商宣称不需要预循环, 但是我们证实预循环大大改善了色谱性能。10 g 树脂混悬于 100 mL 水中, 并用 100 mL 水清洗数次, 每次清洗可去除细小物 (不沉降的小颗粒)。然后用 100 mL0.Imol/LHCl 处理树脂 30 min, 将 HCl 缓慢倒出。至少清洗 3 次树脂, 每次用 100 mL 水。最后一次清洗缓慢倒出液体,然后用 0.Imol/LNaOH 处理树脂 30 min, 缓慢倒出液体,至少清洗 3 次树脂,每次用 100 mL 水。用抗体用缓冲液清洗树脂 3 次,每次用 100 mL 缓冲液, 并用相同的缓冲液悬浮。用 pH 试纸检测 pH,加人叠氮钠至终浓度 0~ 0 2%。树脂分装于一次性试管,存放于冰箱内。
(2)mAb 可来源于腹水。虽然腹水并不纯 (纯度 80%~90%), 但杂质可能并不影响免疫亲和树脂的性能。
(3) 在上述条件下,大多数小鼠 mAb 可流穿 DE52。但仍有少量 mAb 可以结合于层析柱。因此,可以采用高盐洗脱条件 [步骤 (7)] 洗脱抗体。使用盐梯度浓度纯化与 DE52 层析柱结合的 mAb 是必要的。
(4) 如果以 CELLine 培养瓶制备 mAb,可不必采用 DE52。
6.PR-mAb 在层析载体上的固定
供应商提供了很多种树脂和偶联剂。我们已经对其中的大部分进行了验证,但没有任何一种比用溴化氰 (CNBr) 衍化的交联琼脂糖更有效。
(1)纯化后 mAb 用偶联缓冲液透析 (1 00 mm o l/LNaHCO3、500 mmol/LNaCUpH8.3)。移出透析管中的抗体溶液,并用偶联缓冲液调整其体积,每克溴化氰活化的 Seph-arose 干粉对应 10 mL, 留样 (约 1 00^L) 分析蛋白质浓度。
(2)约需 20 min 溴化氰活化的 Sepharose 干粉即可溶胀于 0.Immol/LHC1。每克溴化氰活化树脂干粉可制备 3.5 mL 凝胶。然后在玻璃滤器内清冼树脂,每克树脂用约 100 mL0.Immol/LHCl。
(3) 用约 20 mL 偶联溶液快速清洗树脂后,将树脂转移至抗体溶液中。23°C 条件下,用实验室旋转器将胶浆液翻转混合 2 h。
(4) 用玻璃过滤器收集树脂, 保存滤液用于测定未偶联蛋白质含量。
(5) 将树脂转移至约 1 0 1111^1”[8.3、1111 0 1/1 乙醇胺溶液中,23。(:翻转混合 211, 使乙醇胺与残余的溴化氰充分反应。
(6) 再次用玻璃过滤器收集树脂,偶联缓冲液 (约 50 mL) 清洗后,用 100 mmol/L 乙酸钠缓冲液 (pH4.0) 清洗。
(7) 重复至少 2 次或 3 次步骤 (6) 中的 2 次清洗。
(8)4°C 条件下,将偶联的 Sepharose 保存于 I0 mL 含 0.02%NaN3 偶联缓冲液中。
(9) 检测结合前后保存的抗体溶液 [步骤 (1) 和 (4) 得到的样本] 中的蛋白质含量。据此确定偶联效率。
说明
(1)Ig 树脂干粉制备 3.5 mL 溶胀树脂。我们发现多数情况下,—次处理 0.5~2.0 g 树脂干粉比较方便。并且证实 ImL 溶胀树脂偶联 2.5 mgmAb 效果较好。因此,3.5 mL(lg) 树脂需要结合 8.75 mgmAb。
(2) 溴化氰活化的 pH 条件是 8 以上。因此, 上述步骤 (3) 中的抗体溶液应尽快转移至树脂。
(3) 阻断剂 [步骤 (5)] 可依据具体使用目的而异。例如, 产自酵母的乙醇胺结合蛋白,如果用乙醇胺作为阻断剂,将会与目标蛋白质共纯化。在这种情况下,almol/L 甘氨酸是比较合适的阻断剂。
(4)4°C 条件下,将树脂储存于 0. 0 2%NaN3 中。该条件下,树脂可以稳定储存约 6 个月。我们注意到,一些抗体储存 6 个月后会发生剥离。向上述缓冲液中加入 50% 甘油,存储于 20°C(不冻结),可能延长半衰期。
7.用 PR-mAbs 纯化蛋白质
下面我们以 mAbNT73 或 8RB13 纯化大肠杆菌来源的 RNA 聚合酶为例。前面介绍一步免疫亲和层析法可以得到纯度约 90% 的 RNA 聚合酶。其他一些与 RNA 聚合酶结合的蛋白质也会被共同洗脱。该操作规程设定原料为 IL 对数生长末期培养液获得大肠杆菌沉淀(2~3 g 湿重)。图 28.3 所示 SD^PAGE 结果为利用该方法纯化的结果。
(1) 可以在冰上部分复融沉淀,并用 20 mLTEN 缓冲液 [50 mmol/LTris-HCl(pH7.9)、0.Immol/LEDTA、100 mmol/LNaCl] 重悬。
(2) 添加溶菌酶至终浓度 250pg/mL, 冰上孵育细胞 20 min。或者使用 1500kU 重组溶菌酶 (EMD/Novagen 公司#H110)。
(3) 冰上超声细胞, 重复 4 次,每次工作 15s,间歇 15s。
(4)15000r/min(27000 g) 离心裂解液 15 min。
(5) 于 23°C 将上清液 (1~2 mL) 上样于免疫亲和柱,收集流穿部分。
(6) 用含 100 mmol/LNaCl 的 TE(约 20 mL) 清洗层析柱,然后再用含 500 mmol/L(原文为 500 mL, 译者认为原文笔误)NaCl 的 TE(约 5 mL) 清洗层析柱。用含 100 mmol/LNaCI 的 TE(约 10 mL) 再平衡。
(7) 用含 0.75mol/LNaCl 和 40% 丙二醇的 TE 洗脱层析柱 (室温)。冰上收集洗脱组分。
(8)SDS-PAGE 电泳分洗脱峰。图 28.3 中 SDS-PAGE 结果显示一步层析产物纯度。合并所需组分(图 28.3 泳道
5、泳道 6),用合适的储存缓冲液透析 [转录蛋白,用 50 mmol/LTris-HCl(pH7.9),50
mmol/LNaCUO.Immol/LEDTA.O.lmmol/LDTT 和 20%~50% 甘油]。

说明
(1) 用 Benzonase 核酸酶处理裂解液 [步骤(3)](EMD/Novagen#7 0 746,Madison,WD,有助于消化核酸和降低黏度 (见本书第 18 章)。
(2)500 mmol/LNaCl 清洗 [步骤 (6)] 有助于消除核酸和 NusA, 后者为一种 RNA 聚合酶结合蛋白。
(3) 室温下洗脱目标蛋白质比 4°C 条件下更有效 [步骤 (8)],原因未知。
8.用 PR-mAb 的交叉反应纯化蛋白质
通常可从非基因工程制备的生物材料中纯化蛋白质或蛋白质复合物。并且发现,使用识别高度保守表位的 PR-mAb 可以使免疫吸附的适用性更强。其中两个应用最成功的 PR-mAb 可以与许多物种的同一种酶发生交叉反应。
mAb8WG16 与真核生物 RNAPn(表 28.1) 最大亚基 C 端的重复七肽产生相互作用。这一序列是该大蛋白质复合物表面容易靠近的部分,通常称之为 RNAPII 的 C 端结构域 (CTD)。RNAPII 中的 CTD 对于所有物种几乎都是高度保守的。mAb8WG16 已用于纯化小牛胸腺(Thompsonetal.,1990)、酵母(EdwardsetaL,1990) 和人体细胞 (Maldonadoetal.,1996) 中的 RNAPII。
事实上,PR-mAb 纯化的酵母 RNAPII 第一次应用是在 RNAPII 晶体化研究方面 (Crameretal.,2000)。我们已经介绍了利用 PR-mAb 纯化 RNAPU(ThompsonandBurgess,1996) 的详细过程。在大多数情况下,从原料中纯化 RNAPII 时需要在使用免疫亲和树脂之前先采用直接混合的纯化程序。
mAb8RBI3 是一个具有高度交叉反应的 PR-mAb, 已证实对纯化细菌核 RNA 聚合酶(coreRNAP)
非常有效(Bergendahletal.,2003;Probascoetal.,2007)。这种 PR-mAb 可与大多数细菌来源的 RNA
聚合酶高度保守的「卩-flap」结构域发生反应 (E.S.Stalder, 未公开发表)。是一个与 sigma
亚基结合的主要位点(Kuznedelovetal.,2002),因此可用 8RB13-agarose 层析柱纯化 RNA 聚合酶 (缺少一个
sigma 亚基)。图 28.4 泳道 3 为由大肠杆菌纯化的核 RNA 聚合酶。鉴于该 mAb 的交叉反应,也可以从枯草芽孢杆菌中纯化核
RNA 聚合酶 (泳道 4)。图 28.4 也显示了用 mAbNT73 从大肠杆菌中纯化的 RNA 聚合酶 (泳道 2),这是一种全酶
(holoenzyme) 与核心酶 (coreenzyme) 的混合物, 两种形式酶组分可以用离子交换色谱法分离
(Thompsonetal.,1992)。纯化大肠杆菌和枯草芽孢杆菌来源核 RNA 聚合酶时,与主要的 sigma 因子对应的肽段是缺失的。

9.使用 PR-mAb 的抗原表位作为纯化标签
我们认为,标准杂交瘤技术分离得到的 5%~10%mAb 都可能是有效的 PR-mAb,即使如此,也可使用另一种方法, 即用已确定为 PR-mAb 的 mAb 的表位对蛋白质进行表位标记。纯化标签的方法在第 I6 章中已介绍。因此,我们将只简单地介绍这一内容,并且仅涉及与 PR-mAb 和其抗原表位相关的部分。
可以用表位标记蛋白质,并用 PR-mAb 通过多轻基反应纯化蛋白质,这种能力正是抗体自身特性使然,而不是表位存在的情况下反应条件的特征决定的。我们已经为 3 个 PR-mAb 开发了表位标记系统,分别为 NT73、IIB8、8RB13(DuellmanetaL,2004;Thompsonetal.,2003;E.S.Stalder,未发表资料)。我们称这些表位为「软性标签 (3 此-ags)」(BurgessandThompson,2 00 2)。表 28.1 中已列出这些表位,且这些标签已获ZL授权(Burgessetal.,2007)。
我们所有 PR-mAb 的分离均以全长蛋白质作为免疫原,因此,这类 mAb 表位的定位是一个辛苦的过程。用合成肽作为免疫原分离 PR-mAb 虽然可行但我们并未采用。PR-mAbIIB8PR-—可以与人 TFIffi 反应—是通过定点突变、用噬菌体展示技术定位的 (Duellmanetal.,20 0 4)。而与大肠杆菌 RNA 聚合酶最大亚基反应的两个 PR-mAb(rnAbNT73 和 mAb8RBI3),是先通过有序片段梯度法(orderedfragmentladdermeth-o d) 进行非严格定位 (BurgessetaL,2000;Ra o etal.,1996), 然后通过细微缺失分析及寡核苷酸标记无关蛋白质 (Thompsonetal.,2003;E.S.Stalder, 未发表数据) 的方式精确定位,这种无关蛋白质我们已有相应抗体。这种寡核苷酸标签的构建是将一段寡核苷酸与目标蛋白质基因融合,这个寡核苷酸包含与目标蛋白质读码框相容的、编码表位的序列。
在该技术的原理论证 (proof-of-principle) 研究中,我们以绿色突光蛋白 (GFP) 为目标蛋白质(DuellmanetaL,2004;Thompsonetal.,2003)。mAbNT73 的表位标签可以与 GFP 的 N 端或 C 端融合,但对于其他目标蛋白质,可能取决于其末端的可及度。对于 o..,其晶体结构中两个末端均易靠近 (丁也 11,1998)。111 八匕 81^13 的表位已经用做大肠杆菌和哺乳动物细胞培养系统的标签 (E.S.Stalden 未发表)。
我们已经将大肠杆菌 RNAP 来源的表位标签用于纯化大肠杆菌表达系统产生的表位标记 GFP, 即便如此仍有内源性 RNAP 存在于裂解液中,同时也有一些 RNAP 被纯化。图 28.4 的泳道 5 显示了大肠杆菌表达、NT73-Sepharose 纯化的表位标记 GFP,其中的表位是针对 mAbNT73 表位。将裂解液加至 300 mmol/L 的 NaCU 同时加入 0.3% 聚乙烯亚胺 (PEI),可以去除其中的 RNA 聚合酶 (见本书第 20 章)。可以通过离心去除沉淀,而表位标记 GFP 仍在溶液中,然后将上清液用于免疫亲和层析。
结论
本部分介绍了 PR-mAb 的分离和鉴定,以及其在温和的免疫亲和层析方面的应用。
虽然这里列举的 PR-mAb 应用实例是将其用于纯化转录因子, 但是本章概述的方法可以用于任意一种 PR-mAb 或用某种 PRrmAb 表位标记的蛋白质。该程序中耗费最大的部分是 PR-mAb 分离。现在针对某一种蛋白质的 mAb 非常多,可以利用 ELISA-elution 检测法对现有 mAb 进行筛选,继而用于多羟基反应,因此完全可能已存在一种针对你的目标蛋白质的 PR-mAb。
展开