蛋白质纯度测定实验(二)
方法
制样的方法根据所选用电泳方法的性质而定。读者可参考本卷其他章节中有关非变性凝胶电泳和等电聚焦电泳的讨论。最常用的 SDS 凝胶电泳是蛋白质纯度检测的「第一线」(front-line) 方法。由于通常纯度评价是非定量的,因此不需要关心诸如极端大小分子的非线性迁移、蛋白质修饰造成的迁移异常以及 SDS 结合不均勻等 Rhodes 等 (2009) 讨论过的问题。另外,需要考虑样品处理和制备过程中的一致性和均一性,以及分离过程中样品所处的环境。例如,制备均一的蛋白质样品时,未彻底还原二硫键将会使结果表现出异质性。同样,凝胶交联不均一导致的胶内迁移率差异,也会造成对结果的错误判断,但是适当的重复和对照会使这种可能性降到最低。
通常,实验者并不知道一个样品中所有蛋白质组分的大小,因此很难预测胶的浓度,从而以最佳效果分离一组蛋白质组分。梯度胶 (gradientgel), 也称为分级孔隙胶 (gelsofgradedporosity), 能够覆盖非常宽的分子质大小范围。尽管梯度胶不可能产生最好的特定分离效果,但是它可以覆盖最宽分子质量范围,从而最大可能的鉴定某种杂质。分辨杂质的能力主要取决于所选择的胶浓度梯度范围。梯度的设计应该使目标蛋白质条带处于中间的胶浓度。胶顶端丙烯酰胺的浓度 (低浓度) 应该足够低,从而使大分子质量的杂质能够进入到凝胶基质中。胶底部 (高浓度) 浓度应该足够高,使小分子蛋白质能够保持在凝胶基质中。通常预制梯度胶的最低浓度是 4% 丙烯酰胺,高浓度极限标准为 20%。如果这个范围不合适, 可以制备低至 2%、高至 40% 浓度范围的胶。分子质量大约 IMDa 的蛋白质应该能够穿过 2% 的凝胶基质。通常大多数梯度胶的丙烯酰胺高浓度不超过 30%。即使在长时间电泳中也仅有非常少数的多肽能够穿透 30% 凝胶。梯度胶的浓度范围决定着胶的分辨率。因此,虽然 2%~30% 的梯度胶能够检测最宽范围的分子大小,但是在分离分子质量几乎相同的两种蛋白质时,分辨效果却不如 8%~16% 的梯度胶。
梯度的选择很大程度上取决于研究者对整个待测系统的了解。例如,如果不存在特别小和特别大的分子,则最好选择窄的浓度梯度范围的胶或浓度恒定的胶。
如果需要特殊的梯度,制作梯度胶的过程与制作常规聚丙烯酰胺凝胶的过程差距甚微,与用于沉降的蔗糖梯度制作过程也很类似。与常规聚丙烯酰胺凝胶制备过程相比,梯度胶制备难点在于两块不同丙烯酰胺浓度的胶必须平行制作。而比蔗糖梯度制作麻烦是因为梯度胶有丙烯酰胺聚合的时间限制。目前已有一些商业化梯度形成仪,而具体技术方法上的不同可以通过査找相关文献来了解。
梯度胶的电泳过程类似于常规电泳法 [Rhodes 等 (2009) 及本书中其他部分介绍]。
胶分析所用的染色手段由预期污染物决定。可以通过将其他方法与电泳法结合来完成二维凝胶电泳分析,由此增强电泳方法的潜在灵敏度及其获得的信息。一般第二维是梯度 SDS~PAGE, 而第一维可能是非变性凝胶电泳、等电聚焦 (isoelectricfocusinggel) 或者不同条件下的变性电泳 (如没有二硫键还原)。
等电聚焦也能检测宽泛范围的污染物,而且可以与电泳方法联用。商业化预制凝胶在同一块胶上的等电点覆盖范围可达 3~10。等电聚焦步骤见本书其他章节,这里不复述。等电点覆盖范围广是一个相当大的优势,而对于等电点相差细微的蛋白质, 可以通过减小等电点范围 (如 4~5、5~8) 来提高区分的灵敏度。除了能够分离等电点不同的分子,等电聚焦法还可通过其他性质来分辨污染物。
问题和局限
鉴于凝胶电泳法的简单和低成本,通常将其作为纯度评价的首选方法。然而,在采用该方法时也需要谨记一些潜在的问题。对于变性凝胶, 可能出现假阴性和假阳性现象。
如果发生杂质共迁移 (comigrating) 或者杂质无法进入凝胶的情况,则会出现假阴性。因此应该将整块胶,包括浓缩胶和分离胶都染色,并且需要检验蛋白质染料是否合适。例如,样品条带若残留于上样孔或处于浓缩胶和分离胶之间,表明存在高分子质量的杂质,或者杂质溶解度有限。同时,常用的蛋白质染色剂(proteinstain), 如考马斯亮蓝与纤维状蛋白质及糖蛋白的结合力较弱,如此将低估此类杂质的含量。而假阳性的产生, 可能是由于样品制备时发生共价修饰,或者胶不均一和氧化剂残留。非变性凝胶中也会出现类似问题,此外目标蛋白质或杂质净电荷的不确定性也会导致意外发生(Rhodesetal.,2009)。如果杂质的静电荷为零或者与目标蛋白质相反,杂质将不会出现在胶上, 除非使用中心上样水平凝胶 (center-loadedhorizontalgel),也可以尽可能加宽非变性凝胶电泳的 pH 范围解决该问题。
等电聚焦相关伪迹 (artifact) 的产生主要是由于用来生成 PH 梯度的多聚两性电解质颗粒与蛋白质相互干扰,造成条带 (一般是弥散的) 在胶上的结果与目标蛋白质的等电点无关。检测这种可能性的一种方法是从中选择一条条带分离出蛋白质,并且用相同的等电聚焦过程分析分离出的蛋白质。如果原结果分布反映了样品中的实际异质性,则分离出的蛋白质只能形成与原始同样的点。然而,如果原始条带是假象,那么分离出的蛋白质将重现原来的分布模式,包括虚假条带。
三、色谱法
1.凝肢过滤色谱法
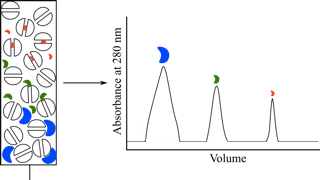
凝胶过滤色谱法是检测与目的分子大小不同的杂质的最简单方法之一。这一方法无破坏性并且非常快速。因为这是一种「区带方法」(zonalmethod), 样品通过凝胶柱时会被稀释, 因此需设置恰好高于最小检测限度的起始浓度。而确切使用量则取决于用本方法检测杂质时的灵敏度。
方法
本卷介绍的测定蛋白质大小的样品和凝胶柱制备的方法, 在本章中将用来评价杂质 (Rhodesetal.,2009)。两种应用唯一的区别在于,后者所用检测方法不仅需要对目标蛋白质的灵敏度高,对杂质同样要求高灵敏度。尽管评价样品纯度时并不需要校准凝胶柱,但这样做有两个好处。首先,实验者使用校准凝胶柱可以获得有关杂质大小的信息;其次,使用校准凝胶柱可以在检测杂质存在的同时测定分子质量。为此, 通常需要进行两个测定: 第一个是蛋白质的非特异测定 (如测 280nm、220mn 吸光度);第二个是特异检测 (酶活检测、免疫学检测、质谱、多角度光散射检测等),用来确认所分析蛋白质的身份。
杂质在色谱图中可能表现为独立的峰,也可能使洗脱谱变宽 [如肩台(shoulder)]。原则上,洗脱谱应该接近高斯分布 (边缘有微小的偏移,见问题和局限)。如果分析蛋白质峰有强烈偏移、峰有肩台,或者特异和非特异测定结果不相符,说明样品中存在杂质。
问题和局限
凝胶过滤色谱法检测分子大小的灵敏度低于电泳法。一般来说, 凝胶过滤实验需要的材料量大于电泳法所需。由于凝胶过滤色谱通常在天然状态下进行,因此结果可以反映样品异质性。如果蛋白质以一系列稳定的寡聚物形式存在 (如脂肪酸合酶),在凝胶过滤色谱将表现出异质性。同样,发生快速、可逆的自联作用的蛋白质会表现出异常的浓度依赖性洗脱谱。这些情况下,可通过进一步测定来区分异质性和分子偶联。例如,从不对称峰和加宽峰中选取不同部分来重复胶过滤色谱法,或采用本章和本卷介绍的其他方法测定选取的部分。这些方法都可以提供目的分子的有用信息。
2.反相高效液相色谱法
另一个应用广泛的色谱分离方法就是反相高效液相色谱法 (reversedphaseHPLQ。
这种方法用诸如改性硅介质等非极性基质作为固定相,进行极性递减的梯度洗脱。例如,在含 0.05% 三氟乙酸 (TFA) 的缓冲液中,蛋白质将通过疏水性氨基酸与 C-18 改性硅介质结合,从而结合于固定相,再从 0.05% 三氟乙酸逐渐提高到一定量合适的有机溶剂洗脱蛋白质, 如乙腈、甲醇或乙醇等,通常三氟乙酸含量保持一致。流动相中的有机溶剂减小了蛋白质与固定相的亲和力,从而将蛋白质洗脱下来。与其他梯度洗脱方法一样,缓慢的梯度洗脱可以获得最佳分辨率,但是首次实验时最好先进行快速梯度洗脱,如此可以得到主要分析物的洗脱最适溶剂条件,并可以根据与固定相亲和力不同来筛选可能的杂质。
蛋白质的检测通常是基于 215~220nm 处的紫外线吸光度来检测肽段,而不是采用 280nm 检测的芳香族氨基酸方法。暂且不论准备时间,梯度洗脱可在 Ih 内完成,大部分优化后的洗脱步骤将在 15~20 min 内完成。
由于该方法简单且快速,具备现成的合适设备,能够灵活应用多种不同特性来分离亲和蛋白质,因此已普遍应用于蛋白质样品的纯度筛选检测。如有必要,可采用多种不同的流动相使样品可以在不同梯度和条件下得到分离,来确定主要分析物的纯度。该方法还可以与其他方法联用 [如质谱法 (见下文)] 来确定蛋白质身份并获得可能存在杂质的信息。值得注意的是,由于有机溶剂的存在,洗脱下来的蛋白质可以预见到有一定程度的变性,不一定能够保持天然构象。使用 C-4 或 C-8 固定相能够将变性可能性降低到最小,这是因为它们与蛋白质有较弱的亲和力,较低浓度的有机流动相就可用来洗脱。许多针对特定蛋白质的操作方案已经发表,HPLC 设备制造商也提供了许多产品信息。
四、沉降速率测定法
沉降速度法 (sedimentationvelocitymethod) 能够简单快速且非破坏性的来评价一个蛋白质的纯度,这种方法对分子质量和分子大小的比值非常灵敏。关于沉降速率测定法在 Rhodes 等 (2009) 的文献中有简要介绍。一般来说,当使用沉降速率测定法来检测蛋白质纯度时,实验者能够找到的沉降组分不止一种。该方法的优点在于测定的材料范围非常广 (尤其是使用折射式光学系统时),但最大的局限在于,它对分子质量相差小的样品的灵敏度不如电泳技术,甚至区带沉降 (bandsedimentation) 也不可避免这种问题。
方法
本卷中 (Rhodesetal.,2009;Coleetal.,2008) 详细介绍了除区带沉降法之外的一些方法, 如样品制备、选择合适的光学系统及实验结果的分析。如何准备离心来实施速率区带沉降实验 (rate-zonalsedimentation) 可参考设备说明书。利用超速分析离心机来实施区带沉降法的细节可以参考 Eason(1984) 或 Ralston(1993) 的文献, 数据的分析类似于凝胶过滤色谱法。
差示沉降系数分布(differentialsedimentationcoefficientdistribution),g(s)(Coleetal.,2008) 分析,是非常有效的杂质检测方法。由于这种方法是模型非依赖性的,因此多组分的存在形式将会是不同夂沉降系数) 值的峰,或是代表主要分析物的峰加宽。另外,与凝胶过滤层析法相同,尽管蛋白质纯度很髙,自身结合蛋白质的沉降行为表现为峰加宽或多峰,使用平衡超离心或其他方法可以帮助解决这个问题。
五、质谱法
由于质谱法可以直接测定一个样品中共价质量的分布,因此这种方法能够非常简便、灵敏地测定杂质。质谱不仅可以检测杂质的存在,还可以描述杂质的质量特征,因此质谱法常被用于鉴定杂质的来源。除了能够直接测定蛋白质分子质量大小,通过串联质谱 (MS,’MS 或 MS2) 法,如碰撞诱导解离(collisioninduceddissociation,CID)、电子转移解离(electrontransferdissociation,ETD)、电子捕获解离(electroncapturedissociation,ECD) 及红外多光子解离(infraredmultiphotondissociation,IRMPD) 等,还可以鉴定共价改性修饰特征和位点。其中,CBD 和 IRMPD 可以测定相当小的蛋白质 (<15kDa),而 ETD 和 ECD 可用于分子质量较大的蛋白质 (约 50kDa)。除此之外,共价修饰键改性及其位点的测定可以用任意一种常用的蛋白质水解法,如胰酶或溴化氰(CNBr)(Link,2009)。在使用飞行时间质谱 (time-of-flightmassanalyzer) 分析前,CID 可通过四级杆碰撞反应池 (quadrupolecollisioncell) 实现(Q-toFconfiguration)。ETD、ECD 和 IRMPD 常用于离子讲分析仪(ion-trapanalyzer),如线性离子讲(lineariontrap)、轨道讲(orbitrap)、轨道讲及傅里叶变换离子回旋共振质谱仪(fouriertransformioncyclotronresonancemassspectrometer)Q 与光散射法类似,将质谱法与其他方法联用 (如凝胶分离色谱) 可以达到最佳的效果。
由于杂质可能与主要分析物有相同的质荷比,因此将质谱法与其他分离方法联用可以增加样品纯度测定的准确度。例如, 如果在凝胶排阻层析的洗脱峰中出现不对称峰,质量检测将帮助区分其是由大小不同的杂质所导致,还是由于自身相关的分子所导致。不同于光散射法基于回转半径 rg(theradiusofgyration) 测量而计算质量,质谱法对蛋白质的质量测量更加准确。
六、光散射法
如 Rhod=等 (2009) 所述,目前一些仪器能够进行静态或动态光散射来测定单个样品,或者监控高效液相色谱和场级 (FFF) 分离过程。通过对分子大小和表观分子质量的连续测定,增强了鉴定洗脱蛋白质的能力,能够区别待分析物、待分析物多种形式的聚集体或杂质。光散射法非常简单且无破坏性,并且目前的仪器能够提供一个洗脱时间函数的分子质量图。这样,如果一个紫外线检测峰不对称,多角度光散射 (multipleanglelightscattering,MALS); 也称多角度激光散射 (multipleanglelaserlightscattering,MALLS) 分析结果则可能表现为峰的主要部分的表观分子质量为 mol/L,而边缘为 2mol/L, 这说明可能存在二聚体。需要注意的是,倍数分子质量的存在并不证明一定存在自身结合,还需采用不同的方法验证该结果。尽管 MALS 结果不如质谱准确,但是所需的设备更便宜且操作简单。