XRF问题汇总(八)
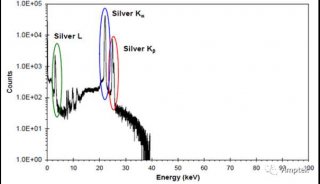
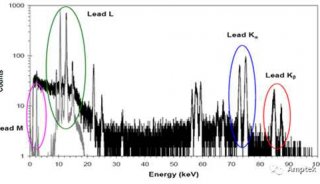
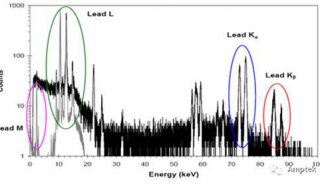
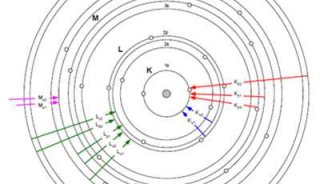
71.铁基中PB的LA线除了因为AS的干扰,还有什么因素干扰?
Pb的La线除了因为As的干扰外,还会受到BiLa或者CrKaSUM的干扰。
72.x荧光光谱报24v故障是怎么回事?
(1) 应该是提供24V的电路板有故障, 测一下24V输出,, 有问题赶快换电路板。 (2) X射线荧光谱仪的各种执行机构驱动方式不外乎电动和气动两种。无论是哪一种驱动方式,上面的电源电压都不会造成故障。引起故障的原因可能有以下几方 面:1、驱动机构自身的问题,如检测动作位置的光电传感器、微动开关、磁性开关工作不可靠造成的位置检测错误;执行元件如微电机、气缸、电磁阀不良造成的 故障。2、控制系统的问题,如印刷线路板控制错误。3、外部干扰造成误动作。4、系统设计不合理。 (3) 检查电路板是不是有虚焊, 我们的衍射仪就出现过这种问题。
73.本人刚刚开始从事X荧光检测工作。(我是冶金行业的)
想请教各位,新进的机器在调试阶段应该注意一些什么问题,厂家来的技术员应该把机器调整到一个什么状态才算调试完成??? 还有就是关于制作检测线的问题,用玻璃熔融方法是不是可以忽略基体效应而不需要校正?玻璃熔融方法制作检测线还有什么需要注意的地方? 如果用压片法,初了基体效应校正还需要注意哪些问题?
厂 家技术员调试仪器会按照厂家的标准逐项进行调试,你可以事先要一份调试标准,做到心中有数.在厂家技术人员来安装之前,你最好把标准样品准备好,仪器调试 好后,请他教你做工作曲线及如何进行基体校正.玻璃融片法是否要做基体校正要看具体样品种类及助熔剂和样品的比例,一般大比例稀释不需要做基体校正.玻璃 融片法要考虑你的样品是否会对铂金坩埚造成损害.压片法要注意标准样品的细度应和实测样品的细度应该一致,同时,标准样品和实测样品的基体也应接近.当然 X荧光分析要注意的事项还很多,你可以找些书看看.
74.我做钢渣最高熔融到1600摄氏度,我还想往高做又怕温度太高把铂金坩锅熔掉! (1) 可以加熔剂降低钢渣的熔点啊!例如碳酸钠、氟化钠等破坏钢渣的结构。直接熔融肯定是不行的。
(2) 它的熔点是:1800-1900摄氏度。
一般来说,难熔样品加入过氧化钠或偏硼酸锂等可以加速融解,但是在融解时加入一些硝酸铵比较好,对坩埚有保护作用。
(3) 直接做炉渣是不可以的,会把坩埚合金化的
(4) 一般实际操作时不要超过1200度.前面说的一定是铂埚被重金属等合金化了,或是金属铁没有吸干净,钢渣制样时也要注意,振动磨时间不能太长,太长时间铁不容易吸出,另外,注意不要混入碳粉等物质.
(5) 加入一定量的氧化剂氧化试样中的还原性物质;碳可以在预氧化的时候应该可以被处理掉。
75. 我是一家公司的品质管理人员,为了应对ROHS指令,在用XRF对供应商的来料进行分析时,发现所测的Pb,Cd的偏差很大,大概在30%左右,但往往发 生尴尬的现象,就是我们测试的不合格,客户提供的报告却合格,这给我们的工作带来不便。我们知道测金属中的元素,往往偏差大,但我希望各位使用过XRF的 经验同仁,能否提供下如何去校正才能使测试结果更有效。
(1) 利用XRF测试误差是必然的,偏差只能减小,不能避免,它基本上属于半定量分析,另外XRF软体资料一般是以常见的塑胶样品建立起来的,当然也有少部分的 金属样品,但这些都远远不能满足实际使用的各种材料,这也是引起偏差的主要原因,还有就是XRF测试依赖原子激发放射光谱,这样的话不能使测试样品数据均 一化,有偏差,另外目前所存在的元素,有很多激发能量比较接近,所以这也会带来很大影响.
(2) 样品中测试元素的特征谱线的强度会受到其他元素的干扰.典型的干扰有 Cd:可能的干扰来自溴,铅,锡,锑
(3) 铅会受溴和砷的干扰,As干扰PbLa Br干扰PbLb
(4) XRF没有自己的方法来做校正,误差很大,但如果校正的好,误差又可以减到很小。我是第三方检测机构的,我们的XRF就做的很好,值得借鉴!
(5) 在使用XRF作检测时,我们自己要注意一点,它只是用来做筛选用的,而且只是测试样品一定厚度的表层,这一结果不能和ICP等定量分析结果相比较。当然,如果样品时比较均质的材质,一般XRF在做筛选时只要调整好其参数,用其来分析的无标线结果也很不错的。
76.我们公司经常在同济大学的x射线荧光分析测量玻璃样品的组分含量.采用的是半定量分析.但是测量出的数据有时有明显出入.请问熟悉x射线荧光分析的大侠们,半定量分析的准确度大概多少?
另外,问过硅酸盐研究所的x射线荧光分析室.他们称Na以下的元素无法用半定量分析出.为什么同济大学可以测量B的含量呢?
(1) 目前的XRF的激发源很难激发Na以下的非金属元素
(2) 半定量分析的准确度不高,并且只适用于判定金属材料中的各元素含量的多少,对有玻璃样品,我个人认为这样的测试方法都不正确更不用说准确度了。
(3) 半定量分析一般都是厂家订好的程序,一般的Be,B,C,需要单独的晶体,不是仪器的标配,要单独订购,并且灵敏度不高。半定量分析时如果预知样品含有大量的B或C要手动输入其值作为平衡相,并且玻璃要做厚度校正,因为它易被荧光穿透。
(4) 现在的荧光光谱仪能够测量F-U的所有元素,但为了延长X光管的使用寿命,通常是很少测量Na以下的超轻元素
77.高频熔融玻璃,高频一般是自动完成的,那么在脱模剂在什么时候添加比较合适? (1) 脱模剂在称样的时候可以加,按1:2的量加的,在熔融摇动时也可以加,但是这时加的量比较少.我们单位开始加的是硝酸锂,在摇动时加的是碘化铵!
(2) 理学的熔融温度一般不超过1250℃。前段时间做了几个熔片,SiO2和Al2O3的混合物,按1:2加无水四硼酸锂不行(使用碘化铵作脱模剂),可以成 熔片,但是倒不出来,后来加到1:4的比例才好烧。一般把NH4I配成溶液,加20~30mg左右,熔融效果比较好。
(3) 在熔样前就可以在坩埚中加NH4I的溶液,在加入样品与四硼酸锂的混合物。据我了解,在做地矿样品时,如果助熔剂的比例较高(一般在1:4以上),可以不 用加脱模剂了,待烧成后直接就可以倒的出来。用马弗炉我也烧过,气泡真的太难消除了,那怕搅拌都存在少量的锅底泡,升高温度能除掉跑,可是到1500℃以 上后有些成分又跑掉了。
(4) 脱模剂加的少,可以脱模。实际影响脱模的原因是,白金坩埚的设计存在缺陷——基本上直角型设计。如果在最后冷却的时候,手工摇动,可以顺利脱模。所以,加入量要多,是我的心里感觉。
熔融时间对熔剂的挥发有很大影响,进而影响主成分含量。
(5) 我个人认为,脱膜主要是和熔融状态下熔融物的粘度有关,粘度小,流动性好,易于脱膜。假如的溶剂主要是考虑被测中的酸碱度,主要是降低熔融物的粘度,加入脱膜剂主要是改变熔融物的离子结果,使聚合的复杂离子变为简单的离子降低黏度,易于脱膜。
加入硝酸氨等物质第一是起氧化作用,第二是生成气泡起搅动作用,有利于混匀样品。 样品里的气泡难于赶出,是因为熔融物内的气泡上浮需要的力和黏度大小有关,黏度大不利于气泡益出。气泡的上浮和湿润角、表面张力、蒸汽压有很大关系
78.我想求助X-荧光压片法做铝土矿的分析方法?
做铝土矿的方法很多文献已经报道过。如果是一个矿山的样品,选择一定梯度的样品先用化学法定值,用基本参数法(需要标样较小,一般选高中低3-6个样品就够了)或a系数法+经验系数法建方法(需要样品多,但效果好)。两种方法可以控制在X%+0.5%(-0.5%)
79.采用真空条件和氦气置换,测出来有硫,而且含量小于1%,但是这样的浓度很难判断究竟有没有硫,那位有招指点一下我只要大致的判断一下含量。
首 先,你要判定是否有S,你要看在你的测试结果里面的图谱,看S的峰对准了没有,如果是对得很准,那就证明有S,而对于量的多少,你要首先确认你的样品的种 类,如果里面是NA之前的元素较多,那么你的这个S的结果的量的可信度就不大了,如果是金属基体,那么你再试一下为S加一个滤波器去测,如果没记错,选用 AL这个滤光片吧,然后按照FP的方法去设,这样测出来的结果比较可信!
80.定量分析里塑胶和铜合金都有快速分析和精确分析,这两者之间是否有区别呢?测试结果一样吗?
(1) 快速分析的时间短,精确分析的结果更可信
(2) 你所说的快速分析和精确分析应该只是测试每个元素的时间不相同,快速分析的时间比较短,精确分析的时间比较长,而每种元素所需要的最短测试时间都不太一 样,所以如果你的快速分析里面的时间可以满足这个要求的话就不会有大的误差,不然误差就比较大。一般进行定量分析时,金属的最低的设置时间是100S(每 种)。小于这个数值重现性不好,误差很大。你可以确认一下这个时间来参考。
(3) 快速分析的意思就相当于远远的看看那个位置有没有元素峰的存在以及预估这个峰的高度
精确分析是准确地把这个峰的高度量出来而后与标准峰比较 可以说定性和半定量以及定量之间的关系。
至于时间不是固定的,只是一个边际的效应问题。
(4) 对于积分测量来说测量时间越长,重现性会越好,但是测量时间也可以更具你的具体要求来设定,只要满足你的测量要求,尽可能的缩短分析测量时间。 金属分析相对来说比较容易,测量时间可以短些
Rosh测量是一个很特殊的领域。一般来说没有必要很长时间,可以设定两种条件,一般情况使用短时间测量,当测量结果位于警戒线附近的时候再用长时间方式测一次,确保准确
-
仪器推荐

-
仪器推荐

-
仪器推荐

-
仪器推荐

-
仪器推荐
 询底价 Tel:400-6699-117 转 7855
询底价 Tel:400-6699-117 转 7855