关于蛋白的处理(二)
G:不知道具体的结合位点?
A:我就选中的原来配体设定的半径。
H:原来的配体就是活性位点,一般半径设置10左右吧,半径太大的话对接时间会长,另外结果不准确,当然半径太小会对接不进去,可以适当调整。
A:你好,我看教程也是说半径设为10但是10的话我看无法全部包括原来的配体分子啊。
H:你可以适当增大,12也可以。
I:你们用的什么软件?我之前对接一个蛋白,用的原配体口袋,对接我的化合物打分挺高的,可是作用的氨基酸跟文献差好多,这正常么?
A:ds,我主要是现在想要选一个好的晶体,把自带的配体对接回去和本身的配体重叠查看一下rmsd值。
G:就是对接出来的结果不是那么准确吧。
A:好多晶体对接回去都不好,我现在调大点半径试试,先对上再说。
I:我之前用薛定谔对接的效果不错。
A:我感觉你那种情况好像也挺正常,这种对接也不怎么准,尤其是ds。
I:后来用的DS对接效果一般,但是作用的蛋白跟我用薛定谔对接的差不多。
A:薛定谔好像是更靠谱一些。
I:做起来挺慢的。
A:你们有薛定谔的教程吗?
I:我是跟别人学了两天,也没太搞明白。原来学校有个课题组做这个方向的,我毕业走了,我们学校开始开的计算机辅助药物设计这门课了!
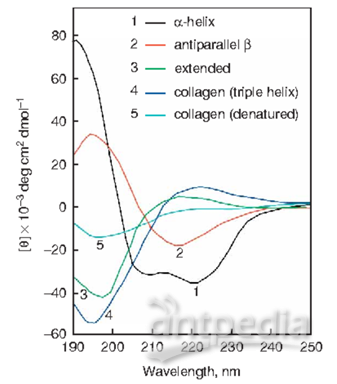
QSAR叠合构象
R:请问,做QSAR分子叠合的时候,有些叠合的不好该怎么搞呢,明明都差不多。殷赋科技:你指的是QSAR模型的预测能力不好吧?如果叠合不整齐,手动调整咯。
R:你是说调训练测试集嘛?
殷赋科技:你是说结构叠合得不好(不整齐)吗?还是叠合后做出来的模型预测能力不好?
R:叠合出来的预测能力还行 confa的0.6几,comsia的有0.7几,就是叠合的不整齐。
殷赋科技:截图看如何不整齐。

殷赋科技:你是不是认为有部分结构应该左右反过来叠合才算整齐?
R:反倒是没反。
殷赋科技:那就没问题啊。
G:这是什么软件啊?
殷赋科技:SYBYL。
R:有几个明明就一点差别,但是叠合出来就重合的不好。
殷赋科技:那是构像问题了,它已经尽力了,但叠合是刚性的,构像如此,它也很无奈啊。调整构像呗,可以用你认为合适的某个化合物来构建相似结构的结构,构像就接近啦。
殷赋科技:或者产生大量构像,然后用算法去挑选。我不知道SYBYL里边有没有这个功能。
R:我刚看了教程找到了你说的利用构象搜索去挑选叠合构象@殷赋科技 感谢你的提醒。
虚拟筛选
S:导师让我做虚拟筛选这块儿(学autodock) ,但是没有头绪,我也不怎么会python,请问有这方面的课程么?
殷赋科技:我们博客上有个脚本,可以用用,不过,并不建议新手去用脚本做筛选。因为计算不是问题,问题是没有经验的人的命中率很低,浪费的是买化合物做实验的钱。
S:有道理,之前同组的同学用svm筛选过酪氨酸酶抑制剂,酶一点点花了八千多。
殷赋科技:我们正在开发在线方案,联合使用vina和dock6对接,再加上根据以往经验写成的分析筛选脚本,能极大提高命中率。如果不着急,不妨等一等。
D:您说的基于Vina和dock6对接,然后经过分析后得到数据的脚本,这个分析是什么个过程?共同命中被选下来?这个的话脚本不难写,如果这个思路可行的话我也想写写试一试。
殷赋科技:主要是两步,一是根据打分挑选最好的化合物,二是根据结合模式挑选,前者容易实现,后者则比较困难。
D:结合模式的话会把关键氨基酸以及结合对关键氨基酸的影响考虑进去吗?
殷赋科技:对,分为一般相互作用(比比如氢键、pi-pi堆积)和特殊相互作用(针对该体系的关键氨基酸及其作用),还有口袋形状的匹配程度HH。
靶标预测图
G:大家有预测疾病靶点的那种图吗?我写东西想用用。但是找不到啊。
F:举个例子?
K:什么样的图?
殷赋科技 :大概要个网络图吧。
G:对就是网络关系那种。
G:不用显示具体的物质。我就是想用一下这样的图。说明一下预测靶点。
殷赋科技:化合物在中间,周围是潜在的蛋白靶标,远近表示可能性大小。