未来可期-CAR-T细胞疗法的发展及展望(二)
● 细胞因子释放综合征(CRS)
细胞因子释放综合征(Cytokine
releasesyndrome,CRS)也称为细胞因子风暴,它是CAR-T临床应用中最显著的不良反应,严重的CRS出现概率为27%~53%,常发生于接受细胞治疗后6~20天[6,
13]。其发生机制是静脉输入CAR-T细胞后,在短时间内清除肿瘤细胞的同时会产生大量的炎症因子,如细胞因子、趋化因子,这些炎症因子会引起炎症反应导致组织损伤,甚至引起死亡。临床表现主要有发热、乏力、疲劳、心动过速、低血压、毛细血管渗透综合性以及心功能不全、弥漫性血管内凝血等症状[14]。目前关于细胞因子风暴造成的不良反应,解决方案主要有:使用糖皮质激素药物(如氢化可的松)来抑制细胞因子分泌;使用靶向阻断刺激细胞因子信号转导通路的药物(如IL-6受体抑制剂)[15];引入可控性自杀基因(如caspase9或者HSV-TK)作为安全开关,在出现急性毒性时,可以快速诱导T细胞凋亡并逆转GVHD。另外,有研究构建一种默认为关闭状态,只有在调控药物作用下才会开启的“On
Switch”CAR-T细胞,用以控制CAR-T作用的时间和剂量,提高治疗的安全性[16]。还有研究显示,CRS
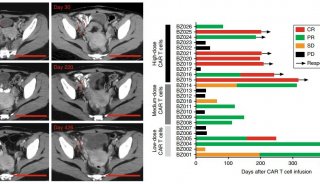
与疾病进程或肿瘤负荷相关,在疾病负荷高的患者体内CRS更严重,因此在疾病恶化之前进行CAR-T回输治疗可降低CRS的风险[6]。
● 脱靶效应
脱靶效应是CAR-T疗法最主要的毒副作用之一。CAR-T细胞与肿瘤相关抗原(tumor-associated antigen,
TAA)具有高度的亲和性,当正常组织与肿瘤细胞都表达同一种抗原时,CAR-T细胞无法区分,在攻击肿瘤细胞的同时会攻击正常细胞。如果靶抗原分子是严格的肿瘤特异性表面分子,就可避免脱靶效应的发生,但真正的肿瘤特异性分子到目前还非常少,因此,寻找仅表达于肿瘤细胞表面的靶抗原是CAR技术应用的首要任务。此外,通过设计多个抗原复合的CAR结构,识别抗原组合,提高杀伤的特异性也可降低脱靶效应的发生概率,比如同时靶向抗原ErbB2和MUC1的CAR-T治疗乳腺癌。肿瘤细胞的基因常常不稳定,一旦靶抗原丢失就会逃离CAR的识别,而间质细胞是肿瘤微环境的重要组分,直接影响肿瘤细胞,且基因表达相对稳定,故有研究选择基质细胞作为CAR杀伤对象,间接作用肿瘤细胞,以应对肿瘤细胞基因不稳定突变。
● 神经系统毒性
接受CAR-T治疗的B-ALL患者中29%~43%可发生神经系统不良反应[17]。CAR-T治疗白血病也可能引起神经系统症状。临床研究报道这种神经系统毒性表现多样,如谵妄、嗜睡、语言障碍、运动障碍、不同程度的脑病症状和癫痫发作[2,
18]。Maude等在接受CAR-T治疗的B-ALL患者脑脊液中发现了CAR-T,提示神经毒性可能与患者脑脊液中出现CAR-T细胞相关[2]。此外,JUNO公司进行的JCAR015Ⅱ期临床试验中,
在20余例患者中先后有3例出现因神经毒性作用导致的死亡事件;分析认为患者可能存在白血病细胞入侵中枢神经系统造成中枢神经系统白血病(Central
nervous system
leukemia,CNSL),导致CAR-T细胞进入中枢神经系统杀伤肿瘤细胞,大量肿瘤细胞短时间内在脑组织中迅速裂解,从而形成脑部水肿;
因此在临床治疗中应排除CNSL患者。CAR-T治疗引起的神经毒性症状通常是自现性的,多数患者神经系统障碍可在短期内恢复,且没有后遗症。使用妥珠单抗或皮质类固醇激素无法明显缓解神经系统的症状,其背后的发生机制未明。
● 插入突变
CAR-T技术是在T细胞中插入一段外源的DNA片段,理论上说其结构已被破坏,存在一定的致瘤风险。虽然目前并没有关于基因改造T细胞致瘤性的报道,但研究者还是一直关注此问题的发生,也在不断通过优化载体的类型和转染的方式来降低插入突变的风险,某些研究中心已采用mRNA直接转导T细胞的方法来避免风险的发生[19]。此外,CAR-T疗法存在其它的不良反应,包括:过敏反应,移植物康宿主病(GVHD),肿瘤溶解综合征等。
4.CAR-T细胞疗法未来发展趋势
一是治疗效果待提高。仅从短时间内的治疗效果看,CAR-T疗法对某些血液肿瘤的治疗效果十分显著,但若将观察期限进一步延长则发现,许多患者在经过CAR-T治疗几个月或更长时间后,会出现肿瘤复发的现象,影响患者预后。因此提高CAR-T细胞激活程度,提高其增殖能力,并延长CAR-T细胞在患者体内的存活时间是未来研究的重点方向之一。针对经CAR-T治疗后血液肿瘤可能再次复发的风险,我们可以设计同时靶向CD19与CD22分子的双特异性CAR-T,已有研究发现,在CAR-T治疗过程中,一些原本表达CD19的癌细胞可能转而表达CD22蛋白,因此CD19、CD22双管齐下,理论上能够增强CAR-T的效果。当前,改善CAR-T疗法在实体瘤中的治疗效果面临巨大的阻碍。
二是CAR-T的产业化。CAR-T疗法费用昂贵。目前,已获批的CAR-T产品和绝大多数CAR-T临床试验都是使用自体型CAR-T,即以患者自身的T细胞为起始材料,这种“私人订制”疗法的生产成本较高。诺华、Kite
获批的CAR-T
产品定价分别为47.5万美元、37.3万美元,高昂的价格限制了其市场潜力。诺华旗下的CAR-T产品在2018年第一季度取得了1200万美元的营收,只达到了预期的三成;Kite的CAR-T产品在获批后两个月内的销售额也不及预期。除费用高昂外,CAR-T疗法质量稳定性也比较差。患者自身的T细胞通常都存在质量与数量的缺陷,不同批次产品之间质量的稳定性较差,难以达到大规模的工业化生产及标准化的质量控制。
三是降低CAR-T细胞毒性。CAR-T疗法表现出的毒副作用说明,需要开发一些控制程序来调控CAR的活性。目前已经有大量方法用于控CAR-T的安全性,比如通过安装自杀开关对移植细胞进行快速清除,这种开关可通过小分子或者抗体来控制。常用的自杀开关包括可诱导的caspase-9(iCasp9)、单纯疱疹病毒中的胸苷激酶(HSV-TK)及自杀表位等。但自杀开关也会清除治疗性的CAR-T细胞,引发治疗反应的不可逆终止。因此,不清除CAR-T细胞的非细胞毒性的可逆系统对于控制毒性反应也许是有用的。
四是开发通用型CAR-T。目前全球主要有十几家公司从事通用型CAR-T开发。该技术主要依赖于基因编辑平台,使用CRISPR技术敲除T细胞中引起免疫排斥的基因,将其制成没有免疫排斥的、只特异性杀伤肿瘤细胞的标准化T细胞,使CAR-T转变为“off-the-shelf”的药品,与高成本“私人定制”的CAR-T不同,通用型CAR-T将会具备能规模化生产、剂量恒定、疗效优越等优点。目前,虽然大多数通用CAR-T的研发还处在临床前或临床早期阶段,但其极具吸引力的治疗潜力足以作为继续研发的强大推动力。通用型CAR-T的上市将会大大降低CAR-T疗法的成本,使这项技术惠及更多的病人。
CAR-T细胞疗法是一直免疫细胞治疗领域的焦点,极有希望彻底改变某些肿瘤(特别是血液肿瘤)的临床治疗现状,美国是全球最早开启CAR-T细胞治疗临床实验的国家,2010年以前,全球CA-T细胞治疗临床注册实验都集中在美国开展,我国从2012年起开始了CAR-T细胞治疗临床注册,每年新注册的CAR-T项目以数倍的速度爆发式增长,目前已成为全球CAR-T细胞疗法临床实验注册数量最多的国家,但是CAR-T细胞疗法在技术上和临床应用上仍然存在着许多亟待解决的问题。希望通过医药企业、临床医生、政府监管部门三方共同努力,建立高水准的标准化治疗流程以及制定一系列的法规政策,推动我国CAR-T细胞治疗技术临床应用的快速发展,为医学发展和人类健康做出贡献。

图3:CAR-T生产过程
5.CAR-T研究相关产品
1)CAR-T活化:CD3和CD28激发型抗体

2)CAR-T扩增:无动物成分重组细胞因子

3)CAR-T鉴定:预包被的ELISA试剂盒

参考文献
1. Goker, H., et al., Chimeric antigen receptor T cell treatment in hematologic malignancies. Transfusion and apheresis science : official journal of the World Apheresis Association : official journal of the European Society for Haemapheresis, 2016. 54(1): p. 35-40.
2. Lee, D.W., et al., T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet, 2015. 385(9967): p. 517-528.
3. Anurathapan, U., et al., Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Molecular therapy : the journal of the American Society of Gene Therapy, 2014. 22(3): p. 623-633.
4. Gross, G., T. Waks, and Z. Eshhar, Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proceedings of the National Academy of Sciences of the United States of America, 1989. 86(24): p. 10024-8.
5. Brentjens, R.J., et al., CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med, 2013. 5(177): p. 177ra38.
6. Brentjens, R.J., et al., Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood, 2011. 118(18): p. 4817-28.
7. Carpenito, C., et al., Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proceedings of the National Academy of Sciences of the United States of America, 2009. 106(9): p. 3360-5.
8. Zhao, Z., et al., Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer cell, 2015. 28(4): p. 415-428.
9. Beatty, G.L., et al., Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res, 2014. 2(2): p. 112-20.
10. Tong, Z.J., et al., Expression and prognostic value of HER-2/neu in primary breast cancer with sentinel lymph node metastasis. Bioscience reports, 2017. 37(4).
11. Kanagawa, N., et al., Tumor vessel-injuring ability improves antitumor effect of cytotoxic T lymphocytes in adoptive immunotherapy. Cancer gene therapy, 2013. 20(1): p. 57-64.
12. Chinnasamy, D., et al., Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. The Journal of clinical investigation, 2010. 120(11): p. 3953-68.
13. Tasian, S.K. and R.A. Gardner, CD19-redirected chimeric antigen receptor-modified T cells: a promising immunotherapy for children and adults with B-cell acute lymphoblastic leukemia (ALL). Ther Adv Hematol, 2015. 6(5): p. 228-41.
14. Kochenderfer, J.N., et al., B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood, 2012. 119(12): p. 2709-20.
15. Maude, S.L., et al., Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England journal of medicine, 2014. 371(16): p. 1507-17.
16. Di Stasi, A., et al., Inducible apoptosis as a safety switch for adoptive cell therapy. The New England journal of medicine, 2011. 365(18): p. 1673-83.
17. Davila, M.L., et al., Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med, 2014. 6(224): p. 224ra25.
18. Turtle, C.J., et al., CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. The Journal of clinical investigation, 2016. 126(6): p. 2123-38.
19. Barrett, D.M., et al., Treatment of advanced leukemia in mice with mRNA engineered T cells. Human gene therapy, 2011. 22(12): p. 1575-86.