







溶出曲线剖析药品内在品质
溶出度系列报道1中曾经阐述过:“多pH值溶出曲线”是固体制剂内在品质呈现在外“一种映射、一种载体”。本期将详细介绍“如何采用溶出曲线‘剖析与肢解’固体制剂内在品质”以及 “如何绘制溶出曲线”。
溶出介质的选用
普通制剂
(1)酸性药物制剂,pH值分别为1.0或1.2、5.5~6.5、6.8~7.5和水;(2)中性或碱性药物/包衣制剂,pH值分别为1.0或1.2、3.0~5.0、6.8和水;(3)难溶性药物制剂,pH值分别为1.0或1.2、4.0~4.5、6.8和水;(4)肠溶制剂,pH值分别为1.0或1.2、6.0、6.8和水。
缓(控)释制剂
pH值分别为1.0或1.2、3.0~5.0、6.8~7.5和水。
解释与说明
(1) 药物pKa值小于3.0的可看作酸性药物,大于等于3.0的可看作中/碱性药物。
(2) 以上pH值之所以给出范围或选项,系因各国要求不同所致,研究者可酌情予以考虑。
(3) 以上含有pH值范围的,可分别按0.5或1.0间隔测试,如溶出曲线差异较大(即pH值依赖性制剂),应分别予以关注;如无明显差异(即pH值非依赖性制剂),美国一般采用1.0、4.5、6.8和水;日本一般采用1.2、4.0、6.8和水。
(4) 如该药物pKa±1.0值未能涵盖于以上各pH值中,建议增加pKa±1.0值溶出曲线的测定,以更好地把握该制剂的溶出特性。
(5) 无论何种制剂都不建议采用pH8.0以上的介质进行表达;如确有必要,应提供充足理由。FDA公布的数据库中,就有“阿维A胶囊”采用pH9.6溶出介质的特例。
(6) 以前人们在进行溶出度研究时,为模拟人体胃液与肠液,还有意识地研究在“含有胃蛋白酶的模拟胃液”和“含有胰酶的模拟肠液”中的溶出情况。
但现今,鉴于所取用的酶给测定结果带来的不确定性与不稳定性,已基本上取消这种研究;取而代之的使用该配制方法,但不含有其中的酶,如FDA公布的数据库中,就有数个品种采用了“SGF(SIF) without enzyme”。
(7) 根据实际需要,可在溶出介质中添加表面活性剂,但应对添加种类与浓度进行评价。浓度研究应从0.01%(w/v)起点、按照1、2、5级别逐步增加,不建议采用3.0%以上的高浓度;且应注意不同来源的表面活性剂可能会对试验结果带来显著性差异的情况,尤其是使用十二烷基硫酸钠时,目前我国中检所已有该试剂的相应标准物质出售。
至于种类,日本倾向采用吐温-80的原因如下:一,药物较易与十二烷基硫酸钠相互作用,对试验结果带来影响,而吐温-80作用较少;二,吐温-80一般不会因来源不同而对试验结果有显著性影响。
(8) 禁止使用有机溶剂,原因有二,一,患者是不可能“先喝二两白酒再吃药”的;二,如该制剂在体内吸收良好,在体外放宽溶出度试验参数时、直至要放宽至“使用一定量有机溶剂的程度”,这样的(创新药物)制剂就值得商榷是否适合做成人体药物了。所以,国外现已规定一般情况下必须革除有机溶剂。
(9) 试验前应首先进行原料药在各pH值溶出介质中的稳定性考察,以确保试验数据的准确测定。在日本橙皮书中,就罗列了该药物在各种溶出介质中的稳定性数据。
溶出介质的配制
欧洲药典配制法
(1)pH值1.0~2.2的盐酸溶液
pH值1.0:精密量取盐酸溶液9.0ml,加水稀释至1000ml,摇匀,即得。
其他pH值溶液:量取一定体积的0.2mol/L盐酸液(量取盐酸18.0ml,加水稀释至1000ml,摇匀,即得),加水稀释至200ml,摇匀,即得。
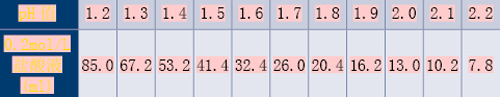
(2)pH值3.8~5.8的醋酸盐缓冲液
2mol/L醋酸溶液:取120.0g冰醋酸(冰醋酸密度为1.049g/ml,故体积约为114ml),加水稀释至1000ml,摇匀,即得。
取下表中规定物质各量,加水溶解并稀释至1000ml,摇匀,即得表2。
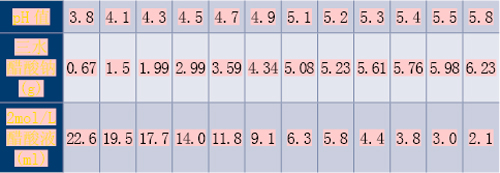
(3)pH值4.5~8.0的磷酸盐缓冲液
取6.80g磷酸二氢钾,加一定体积0.2mol/L氢氧化钠液(取8.0g氢氧化钠,加水溶解并稀释至1000ml,即得)和适量水溶解后,再加水稀释至1000ml,摇匀,即得。
美国药典配制法
(1)pH值1.0~2.2的盐酸/氯化钾溶液
同以上欧洲药典配制,但其中要溶解入0.75g氯化钾。
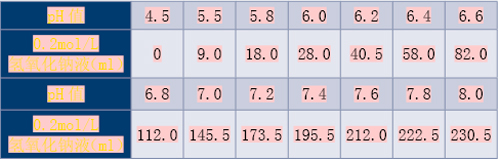
(2)pH值2.2~4.0的盐酸/苯二甲酸氢钾溶液
称取2.04g苯二甲酸氢钾,加入一定体积0.2mol/L盐酸液和适量水溶解后,再加水稀释至200ml,摇匀,即得。
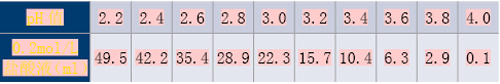
(3)pH值4.2~5.8的氢氧化钠/苯二甲酸氢钾溶液
称取2.04g苯二甲酸氢钾,加入一定体积的0.2mol/L氢氧化钠溶液和适量水溶解后,再加水稀释至200ml,摇匀,即得。
(4)pH值5.8~8.0 氢氧化钠/磷酸二氢钾溶液
同以上欧洲药典配制。
(5)pH值8.0~10.0的氢氧化钠/氯化钾/硼酸溶液
称取0.75g氯化钾与0.62g硼酸,加入一定体积的0.2mol/L氢氧化钠溶液和适量水,溶解后再加水稀释至200ml,摇匀,即得。
日本药典配制法
(1)pH =1.2溶液:取氯化钠2.0g,加水适量使溶解,加盐酸7ml,再加水稀释至1000ml,混匀,即得。
(2) pH值3.0~6.0介质:取十二水合磷酸氢二钠17.91g,加水溶解并稀释至1000ml,即得0.05mol/L的磷酸氢二钠溶液;另取一水合柠檬酸(亦称“枸橼酸”)5.25g,加水溶解并稀释至1000ml,即得0.025mol/L的柠檬酸溶液。然后用柠檬酸溶液调节磷酸氢二钠溶液,使最终pH值至目标值即可。
(3) pH = 6.8磷酸盐缓冲液:取磷酸二氢钾1.7g和无水磷酸氢二钠1.775g,加水适量使溶解后,定容至1000ml,即得。
解释与说明
(1)溶出介质中的离子种类与强度(即“浓度”)对于溶出结果的影响依品种而定;研究者可根据自身情况予以研究。
(2)以上各介质配制的pH值误差范围均应在±0.05以内。
溶出曲线的测定
测定时间点
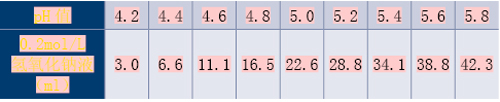
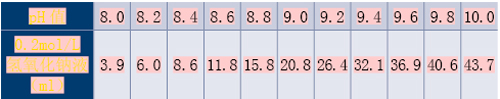
对于测定时间点,普通制剂与肠溶制剂可分别为5、10、15、20、30、45、60、90、120min,此后每隔1h测定;缓/控释制剂可为15、30、45、60、90、120min,3、4、5、6、8、10、12、24h。
结束时间点
对于结束时间点,在酸性介质(pH值1.0~3.0间)中最长测定时间为2h;在其他各pH值介质中普通制剂与肠溶制剂均为6h,缓/控释制剂为24h。但当连续两点溶出率均达90%(缓/控释制剂为85%)、且差值在5%以内时,试验则可提前结束。
解释与说明
(1)测定时间点的拟定:为对固体制剂内在品质进行剖析与肢解,就必须“擘肌分理、抽丝剥茧”般予以分析辨明。很多的研究表明:相当一部分原研制剂的溶出曲线具有延迟、拐点等现象(如辛伐他汀片原研制剂就具有5~10min的延迟释放),这也揭示我们在仿制时应加以注意,因为这些现象对于其后的生物等效性试验具有相当的意义,所以以上测定时间点拟定得较为“紧密”,但当比较测定点确定后,对于其后制剂(如仿制制剂、变更后制剂等)则可仅进行比较时间点的溶出量测定了。
(2)用于溶出曲线比较用试验样品的生产规模:由于固体制剂生物利用度与生产规模密切相关,故一般情况下应不少于今后工业化最大生产规模的1/10或不少于10万个单位。日本规定可选取两者间小的单位;而其他发达国家皆规定不少于10万单位。
(3)比较样品含量差值:用于比较的两种制剂含量差值应在5%以内。
(4)测定数量:为达到统计学要求,规定每个品种的测定皆应选取12个单位。但鉴于目前的溶出仪尚未有12个溶出杯装置,一般测定6个单位即可。众多试验表明:绝大部分原研制剂的均一性与稳定性皆十分良好,这也是目前国际制药行业最为奉行的QbD理念(质量源于设计控制)的体现。
(5)标准溶出曲线范围的确定:在进行仿制药研发时,考虑到原研制剂批间差异与耐受性,建议从市场流通渠道获得有效期内不同时间段的3~5批样品,分别测定后,取结果均值用于比较;并同时确定参比制剂在各pH值溶出曲线的波动范围,以更为有效地评估原研制剂内在质量和自身仿制制剂的研发深入程度。
装置与转速的确定
对于片剂,建议采用桨板法/50转;对于胶囊剂、建议采用转篮法/100转(如采用桨板法、建议采用沉降蓝),系因人们通常认为这两者的机械强度相当。除非特指或特殊制剂,不建议采用更慢转速。
#p#
当在某溶出介质中、结束时间点的最终溶出量未达到90%(缓/控释制剂为85%)时,为便于其后溶出曲线的比较,建议放宽试验参数:桨板法增至75转,转篮法增至120转;如未果,建议添加表面活性剂,如至3.0%浓度仍未果,则再增加至桨板法/100转,转篮法/150转。
累积释放度校正计算公式
在多次取样时、可采取及时补充相同体积同温度溶出介质亦可采取不补液两种方式,但必须保证每次抽取体积的固定性。累积校正计算公式如下:
补液时
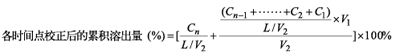
其中Cn为各时间点取出后的样品浓度(即稀释前的);L为制剂标示量(单位需与Cn一致);V1为各时间点固定取样体积;V2为溶出介质体积。
该公式如采用各时间点测得释放量表示,则可演变为:
其中 An为各时间点测得释放量。
不补液时
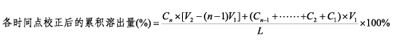
其中 Cn为各时间点取出后的样品浓度(即稀释前的); L为制剂标示量(单位需与Cn一致);V1为各时间点固定取样体积; V2为溶出介质体积。









