基于液质联用的单细胞蛋白质组学研究进展
摘要
蛋白质是细胞功能的主要执行者,由于其无法在体外进行扩增,单细胞蛋白质组学技术相较单细胞基因组学和转录组学技术而言发展相对滞后。传统的蛋白质组学技术可获得大量细胞蛋白表达的平均值,但忽略了细胞亚型及细胞异质性等信息。单细胞水平的蛋白质分析有助于阐明细胞不同表型与异质性的分子基础。随着质谱仪的快速发展,基于质谱的方法将单细胞蛋白质组学推向新的高度。本文综述了近年来基于液质联用方法的单细胞蛋白质组学在单细胞挑选、样品前处理、同位素标签技术、肽段分离、质谱采集、数据分析等方面的研究进展,及其在生物医学研究中的应用,并对未来单细胞蛋白质组学面临的挑战和发展前景进行了展望。单细胞蛋白质组学技术的进步将为生物医学研究领域提供新的思路和解决方案,并加深我们对人类健康和疾病的理解。
前言
蛋白质是生命活动所必须的生物大分子,决定了细胞的结构和活性[1],参与和控制所有的细胞过程[2],是细胞功能的主要执行者。1994年,Wilkins[3] 首次提出蛋白质组的概念,表示基因组所表达的全部蛋白质。作为功能基因组学的重要支柱,蛋白质组学应运而生,其以蛋白质组为研究对象,研究内容包括蛋白质的组成成分、修饰状态、表达变化、蛋白质间相互作用以及蛋白质功能等。随着生物质谱、生物信息学分析等技术的发展,蛋白质组学研究在近十年取得了很大进展,已逐渐成为科学研究中的重要领域,有助于我们从蛋白质水平深入认识生命活动和疾病发生的分子调控机制。
过去几十年,得益于仪器和信息学技术的快速发展,基于质谱的蛋白质组学技术已经能够对人体组织中上万种蛋白质进行深度定量分析,这极大地扩展了我们对细胞信号、调控和代谢通路[4] 的理解。然而,传统蛋白质组学大多是对大量细胞 (大于 106 个) 或组织样本进行分析,得到的是蛋白质表达水平的平均值,忽视了细胞个体对整体的影响,遗失了细胞亚型特异性和细胞异质性的关键信息。因此,传统蛋白质组学在稀有细胞群中的应用受到了很大的限制,如早期胚胎发育、免疫激活、癌细胞的异质性解析等。单细胞蛋白质组学的发展为上述问题的解决提供了新的思路。
早在二十年前,科学家就尝试利用抗体[5,6]、绿色荧光蛋白 (Green Fluorescent Protein,GFP)[7] 或基质辅助激光解析电离飞行时间质谱 (Matrix-Assisted Laser Desorption/Ionization Time-Of-Flight Mass Spectrometry,MALDI-TOF MS)[8] 对单个细胞中的蛋白质进行鉴定和定量。然而抗体的可利用性、特异性及成本等问题限制了单细胞蛋白质分析技术的进展。GFP是一种报告基因,可以分析外源表达蛋白的单细胞分布,分析的蛋白的种类也比较受限。MALDI-TOF MS能在单细胞中鉴定到几十种代谢物、肽段等分子,蛋白鉴定数量较低,定量准确性较差[9]。近年来,基于液质联用的单细胞蛋白质组学技术的快速发展,在蛋白质的鉴定和定量方面均显示出较好的优势。
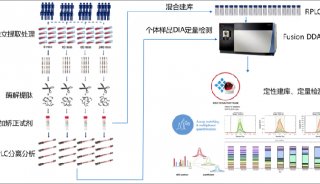
得益于样品制备和实验设计的创新,2018年, Zhu 和 Budnik 等[10,11] 分别报道了哺乳动物单细胞组学的研究成果,优化了基于液质联用的单细胞蛋白质组学技术 (见图1),在单个哺乳动物细胞中鉴定到数百种蛋白质,开启了真单细胞蛋白质组学研究的新纪元。目前,利用无标记定量技术[12] 及同位素标签技术[13-15],单个哺乳动物体细胞中可鉴定一千种以上蛋白质,几百个细胞中可鉴定六千种以上蛋白质[16]。由于单细胞蛋白质组学技术发展时间较短,虽然已被应用于多种细胞和组织研究中,包括单个人体细胞、单个人卵母细胞、显微切割组织等,但仍然处于起步阶段,面临诸多技术挑战。
单个典型的哺乳动物体细胞包含大约 0.2 ng蛋白质,高于当前质谱检测最低检测限,使得单细胞蛋白质组学质谱检测成为可能。样本制备以及进入质谱分析器之前的蛋白分子丢失是制约单细胞蛋白质分析能力的关键因素,也对质谱的检测和数据分析带来了挑战。采用传统蛋白质组学的前处理方式、质谱采集方法和分析策略,在单个细胞中很难获得理想的蛋白质鉴定效果。比如,Wu 等[17] 发现 50 μg 蛋白质样品在经过多次转移后会损失 15%,而 2 μg 蛋白质样品在多次转移后存在 89% 的损失率。可见相同的处理方式对分析痕量蛋白质的影响是无比巨大的。因此,制约单细胞蛋白质分析能力的主要挑战有:(1) 蛋白提取效率和酶切效率; (2) 样品在前处理过程中的吸附损失;(3) 肽段的分离和离子化效率;(4) 质谱采集速度;(5) 数据分析策略。
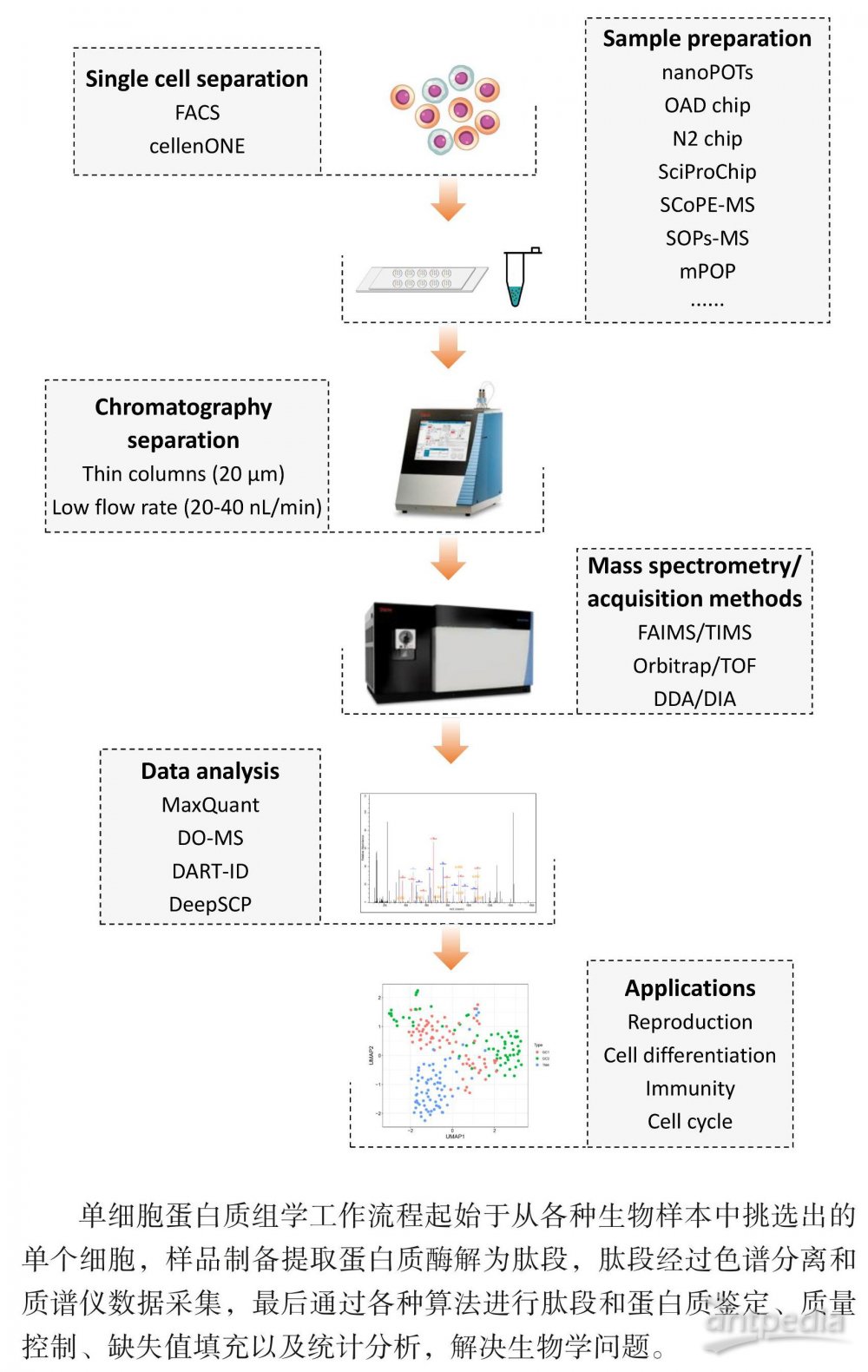
基于质谱的单细胞蛋白质组学技术的突破有赖于常规蛋白质组学流程每个环节的优化,本文将综述以液质联用为核心的单细胞蛋白质组学在单细胞挑选、样品前处理、同位素标签技术、肽段分离、质谱采集、数据分析等方面的研究进展,以及单细胞蛋白质组学在生物医学研究中的应用。
1 单细胞蛋白质组学技术研究进展
1.1 单细胞挑选
生物样本中单个细胞的获取是单细胞蛋白质组学技术开展的前提。目前,单细胞分选技术包括荧光激活细胞分选 (Fluorescence-Activated Cell Sort⁃ ing,FACS)、激光捕获显微切割 (Laser Capture Microdissection,LCM) 和 cellenONE 全自动单细胞分选[18] 等。FACS 最早由 Bonner 等[19] 报道,可根据目标细胞的荧光特性和光散射特性,将其从不同类型的细胞混合物中快速分离,可筛选多达 108 个/ 天,具有高通量[20] 的特点,已被广泛应用于单细胞的分离分析[10],但 FACS 分选过程中的鞘液压力会对细胞活性产生一定的损伤[21]。LCM[22] 是一种利用显微镜根据形态特征识别细胞获得组织细胞亚群的方法,但该技术通量较低,不利于大规模应用。相较而言,cellenONE是一种温和的全自动分选系统,可于数分钟内实现数百个细胞的分离,已应用在单细胞蛋白质组学研究中[23,24]。单细胞挑选技术的不断成熟,尤其是专用自动化仪器的使用,提高了单细胞挑选的准确性和成功率。
1.2 单细胞蛋白质组样品前处理方法
单个哺乳动物体细胞所含的蛋白量低至皮克级[25]。因此,在总蛋白量极低、操作步骤繁琐的情况下,最大限度地减少蛋白质损失,并保证蛋白酶对蛋白质消化效率是单细胞蛋白质组学样品制备的关键。通常采取的策略包括简化样品处理步骤、减少蛋白质吸附损失和最小化样品反应体积。
常规的 “ 鸟枪法 ” 蛋白质组学 (Shotgun Proteomics) 步骤繁琐,其基本流程是从样本 (细胞、组织、体液等) 中提取蛋白质后,使用酶 (如胰蛋白酶) 酶解成肽段,经色谱分离后进行质谱分析。中间涉及到蛋白质还原、烷基化、酶解等多个步骤。倘若样品中含有去垢剂如十二烷基硫酸钠 (Sodium Dodecyl Sulfate,SDS) 或其它与质谱不兼容的试剂,还需要对样品进行纯化处理。因而,在整个样品制备过程中,蛋白质和多肽暴露于各种表面,并由于非特异吸附而导致不同程度的损失。
为了减少样品处理步骤、减少蛋白质吸附损失,研究人员尝试了各种样品前处理装置。Hughes团队[26] 开发了一种基于磁珠的单罐固相增强样品制备 (Single-Pot Solid-Phaseenhanced Sample Prepara⁃ tion,SP3) 方法,将样品制备的所有过程 (细胞裂解、蛋白质酶解、肽标记、纯化、分馏和浓缩) 集中在单个反应容器中完成。该方法兼容大多数去垢剂,在5 ng Hela细胞蛋白提取物中可鉴定到200个蛋白质[27]。同样在单个装置中进行样品制备的还有 iST (in-StageTip)[28] 和 SOPs-MS (Surfactant-Assisted One-pot Sample Preparation Coupled With Mass Spectrometry) 方法[29]。利用 SOPs-MS 方法可在单个细胞中定量到数百种蛋白质,在小组织切片中 (约20个细胞) 定量约1 200种蛋白质。
减少吸附损失对于痕量蛋白或多肽是至关重要的。高浓度的胰蛋白酶酶解[30-33] 可以有效减少酶切产物的非特异吸附问题,但过量胰蛋白酶带来的糜蛋白酶活性会对后续的肽段分析造成干扰[33]。Li 等[34] 利用强疏水性肽段对 0.2 mL 的普通 EP 管和低吸附 EP 管进行包被处理,将单个 Hela 细胞的蛋白质鉴定数量分别提升了 63% 和 23%。最近, Masuda等[35] 开发了一种基于羧基包被磁珠和相转移表面活性剂的油包水 (Water Droplet-in-Oil Diges⁃ tion,WinO)样品前处理方法,可进一步减少蛋白质和肽段的吸附损失。该方法将蛋白质和肽段的回收率提高了10倍,可在单个细胞中定量到462种蛋白质。
样品制备体积小型化,一方面可以降低样品与容器表面的非特异性结合减少样品损失,另一方面可提升蛋白质和蛋白酶浓度以提高蛋白酶的酶切效率[36]。Li 等[37] 开发出了一种油-气-液滴 (Oil-Air-Droplet,OAD) 芯片,可将反应体积缩小至 2 μL。该方法在 100、10 和 1 个 Hela 细胞中分别鉴定到 1 360、192和51个蛋白质。Kelly和Zhu的团队[10] 设计了一种名为 nanoPOTS (nanodroplet Processing in One Pot for Trace Samples) 芯片的样品制备装置,通过在恒温恒湿环境中进行样品制备以最大程度地减少液滴的蒸发,并可以借助机器人实现自动化[38]。nanoPOTS 芯片可以将样品制备体积缩小至 200 nL;结合蛋白质谱分析工具 MaxQuant 的运行间匹配 (Match Between Runs,MBR) 算法,可在 10个与单个Hela细胞中分别鉴定到超过3 000个蛋白质与 670 个蛋白质[39]。引入的自动化装置有利于减少批次效应,大大节省人力成本,提高通量。之后以此为基础改进的N2(Nested nanoPOTS)芯片[23],进一步将反应容器的体积减小至 30 nL,分别在单个 C10、RAW 和 SVEC 细胞中鉴定到 1 735、1 690 和 1 725 个蛋白质。此外,通过增加芯片中样品反应容器数目可有效减少单个芯片的处理时间,同时结合同位素标签技术,可将单细胞通量提高10倍。 Gebreyesus 等[40] 开发了基于微流控芯片的单细胞集成蛋白质组学芯片 (Single-cell integrated Proteomics Chip,SciProChip),结合数据非依赖性采集 (Data-Independent Acquisition,DIA) 模式,可在单个哺乳动物细胞中鉴定到约1 500种蛋白质。
总之,单蛋白质组学样品前处理方法的优化对于减少蛋白质样品损失,提升酶切效率,均有着重要意义。未来高效的自动化样品制备设备研发,样本制备体系的进一步简化和效率优化,有助于进一步提高单细胞定量蛋白的种类,提高单细胞样本之间的制备重复性。
1.3 基于同位素标签标记的载体增强单细胞蛋白质组技术
单个细胞样品的质谱检测时间约为 1-3 小时,极大地限制了单细胞蛋白质组学的高通量分析。 Russell等[41] 率先在单细胞蛋白质组学分析上引入同位素标签 (Tandem Mass Tag,TMT) 技术,借助载体 (Carrier/Boost) 通道的使用,可有效降低处理过程中的蛋白质吸附损失,提升蛋白鉴定数量,并一定程度上解决了单细胞蛋白质组低通量的问题。 Slavov 团队[11] 由此开发了质谱分析单细胞蛋白质组 (Single Cell Proteomics by Mass Spectrometry, SCoPE-MS)技术,实现了在单个细胞中定量一千种蛋白质的可能。Woo 等[23] 将同位素标签技术与 nanoPOTS芯片结合开发出先进的 N2芯片,该芯片在单个细胞中平均可定量到1 500种蛋白质。然而,同位素标签技术仍存在部分缺陷,试剂纯度不足会导致蛋白质定量比例压缩[42],载体通道过多亦会影响细胞定量的准确性[14,43]。基于同位素标签标记的载体策略可以提高单细胞中蛋白质的鉴定数量和通量,但如何解决载体对单细胞蛋白质定量的准确性的影响仍需要进一步研究。
1.4 液相色谱肽段分离方法
肽段的高效分离可降低给定时间内进入质谱仪的样品成分的复杂性,扩大动态范围并减少电离抑制,肽段分离和电离的改善是进行超灵敏质谱分析的重要条件[44]。目前,大多数蛋白质组学分析采用 75 μm 内径的色谱柱和 300 nL/min 左右的流速[45]。研究表明,更细的色谱柱 (如 20 μm 内径色谱柱) 可以将蛋白质质谱鉴定的灵敏度提高 20 倍以上[46],而液相色谱的流速降至20~40 nL/min时,质谱灵敏度亦可获得显著提升[47]。Cong 等[48] 采用 20 μm 内径的色谱柱和 20 nL/min 的色谱流速针对单个 Hela 细胞蛋白质进行质谱鉴定,发现在使用相同的细胞类型、制备方法和质谱仪的条件下,更细的色谱柱与更慢的流速使得平均MS/MS衍生鉴定率增加了40% 以上,达到近300个蛋白质。此外,多孔层空心柱 (Porous Layer Open Tube,PLOT)[49]、窄孔径纳米色谱柱[48]、毛细管电泳 (Capillary Electrophoresis, CE)[50] 皆可以显著提高微量样品中的蛋白质鉴定效果。内径更细的高效液相色谱柱和更低的色谱流速能提升单细胞蛋白质组学的检测灵敏度,但也会带来实验操作难度,延长色谱梯度时间,降低分析通量。开发适用于单细胞样品的高灵敏度新型色谱柱,是未来的发展方向。
1.5 质谱仪及采集方法
质谱性能的提升驱动单细胞蛋白组学的飞速发展。目前绝大多数微量样品的分析都使用了基于 Orbitrap 的高分辨质谱检测器。与 Orbitrap Fusion Lumos 质谱仪相比,最新一代的 Orbitrap Eclipse 可使单个 Hela 细胞中鉴定到的肽段和蛋白分别增加 36% 和 20%[48]。最近,离子淌度 (Ion Mobility) 技术在蛋白质质谱中的应用显著提升单细胞蛋白质的鉴定效率。基于漂移管的离子迁移谱 (Ion MobiIity Spectrometry,IMS) 和高场不对称波形离子迁移谱 (High-Field Asymmetric Waveform Ion Mobility Spectrometry,FAIMS) 可以过滤单电荷物质,提高质谱对肽段的选择性[51,52],以提升单细胞蛋白的鉴定效果[12]。Mann团队[53] 等联合开发的捕集离子淌度质谱 (Trapped Ion Mobility Spectrometry,TIMS) 在微量蛋白和单细胞层面展现了强大功能和广阔的应用前景。
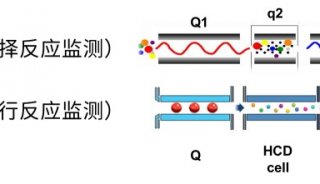
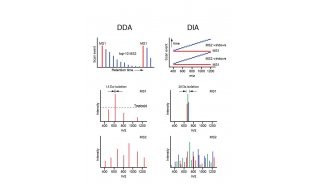
数据依赖采集 (Data-Dependent Acquisition, DDA) 模式是蛋白质组肽段鉴定的最常用方法[54]。在该模式下,质谱会选择强度最高的N个离子 (一般10~20个) 进行碎裂并采集二级谱图。这种方法受限于质谱仪扫描速度和检测灵敏度[55],不能保证每个前体离子在每次运行中都碎片化,从而在肽段和蛋白质鉴定中产生显著变异。在单细胞蛋白质组学中,这一缺点被进一步放大,而数据非依赖性采集 (Data-Independent Acquisition,DIA) 模式可以克服这一缺点。在DIA中,整个质谱扫描范围被划分为多个小窗口,这些窗口内的所有前体离子都被反复碎片化[56],从而可以无遗漏、无差异地获得样本中所有离子的信息。相比于DDA,DIA可以鉴定复杂样品中的低丰度蛋白,极大提升鉴定的蛋白质数目[57],也已在单个哺乳动物体细胞的蛋白质质谱鉴定中发挥优势[40]。
虽然现有的质谱仪灵敏度已经能用于单细胞蛋白质的鉴定,但是很多低丰度表达的蛋白质,质谱仪仍然无法检测。未来更高灵敏度质谱仪的研发,能促进单细胞蛋白质组学方法的发展,更好地实现低丰度蛋白质的检测覆盖度。
1.6 单细胞蛋白质组学数据分析方法
对单细胞质谱数据进行分析与优化,能帮助我们优化单细胞蛋白质质谱鉴定参数,提升定量准确性、增加蛋白质的鉴定效果。 DO-MS (Data-Driven Optimization of Mass Spectrometry)[58] 能够可视化质谱数据并帮助确定最佳质谱参数,如洗脱峰采样和污染水平等;通过增加离子积累时间,可使二级质谱分析的离子递送效率提高 370%。 SCPCompanion (Single-Cell Proteomics Companion)[43] 能够快速分析单细胞蛋白质组学数据并对实验和仪器参数进行优化,可快速评估单细胞蛋白质组学数据,推荐仪器和数据分析参数以提高数据质量。 SCeptre (Single Cell Proteomics Readout of Expres⁃ sion)[59] 是用于数据归一化的计算工具,可以有效地对数据进行标准化,实现细胞聚类。以上几种工具被用来进行质量控制以评估数据的准确性和完整性。
DART-ID (Data-Driven Alignment of Retention Times for Identification)[60] 和 IceR[61] 被用于减少单细胞蛋白质组学实验中的缺失值。DART-ID 通过利用肽段保留时间等信息,提高了单细胞蛋白质鉴定的数量,减少了单细胞定量的缺失值。IceR (Ion current Extraction Re-quantification) 使用离子电流信息进行混合肽段鉴定,与其它方法相比,具有更好的定量精度、准确性、可靠性和数据完整性。
DeepSCP[62] 利用深度学习以提高单细胞蛋白质组学的覆盖率,其通过预测保留时间来构建肽段匹配图谱 (Peptide-Spectrum Matches,PSMs) 的一系列特征,结合优化的集成学习模型预测 PSM 标签。通过目标诱饵竞争 (Target-Decoy Competition, TDC) 的方法以可靠地鉴定肽段和蛋白质。作为一种方便、低成本的计算框架,DeepSCP有助于单细胞蛋白质质谱鉴定,并促进单细胞蛋白质组学的未来发展和应用。
目前已有的单细胞蛋白质组学质谱数据的算法、软件能帮助改善单细胞蛋白质组学的覆盖率。然而,单细胞蛋白质组学数据的谱图解析率仍然不高,谱图解析率的提升和硬件提升同样重要,能有力地促进单细胞蛋白质组学的发展。
2 单细胞蛋白质组学应用
目前,单个哺乳动物体细胞可以定量到一千种以上的蛋白质,其分析深度与转录组学相比仍有一定差距。目前的单细胞蛋白质组学应用主要集中在细胞分群、细胞异质性研究等方面,涉及生殖发育、细胞分化、免疫、细胞周期等各个领域。非洲爪蟾的卵母细胞直径约为 1~1.2 mm,肉眼可观察,易于处理,是研究细胞间异质性和胚胎不对称性的理想模型[63],其蛋白质含量远多于哺乳动物细胞。 Garcia 等[64] 使用纳米流体装置,结合 DIA,从非洲爪蟾囊胚单个细胞中检测出 1 650 种蛋白质,发现 2细胞阶段至8细胞阶段差异表达蛋白质数量增加,细胞间异质性增加,并揭示了爪蟾胚胎发育早期的不对称性。
相比之下,哺乳动物卵母细胞直径约为 50~120 μm,人类单个卵细胞的蛋白质含量约为100 ng。 Virant-klun 等[65] 利用 SP3 技术,在单个人类卵母细胞中鉴定出约450个蛋白质,并对卵母细胞成熟过程进行深入探究,有助于进一步解析人类卵母细胞生物学特征、女性不育的分子机理以及胚胎植入前发育等生物学过程。Guo 等[32] 优化了蛋白质裂解和消化条件,对 34 个人体内与体外成熟卵母细胞进行单细胞蛋白质组学研究,鉴定到2 382种蛋白质,揭示了体外成熟卵母细胞具有高度异质性,为评估体外成熟对卵母细胞质量的影响和研究卵母细胞成熟的分子机制提供了新的见解。针对雌性生殖细胞的单细胞蛋白质组学研究有助于阐明雌性生殖细胞成熟、发育的分子机制,帮助我们更好的理解不孕不育的发病机制,为解决人类不育提供理论基础。
单个哺乳动物体细胞蛋白含量约 0.2 ng,进行单细胞蛋白质组学研究具有更大的挑战。Zhu 等[39] 利用 nanoPOTS 芯片技术,成功区分人肺原代细胞中不同细胞类型,筛选鉴定出上皮细胞和间充质细胞的蛋白质标志物。Schoof 等[59] 利用单细胞蛋白组学研究原发性髓系白血病的干细胞分化,对不同分化阶段的细胞进行分群及分化轨迹分析,发现同一细胞类型的蛋白表达呈现异质性,为细胞发育及分化奠定分子基础。Schulte-Schrepping等[66] 利用单细胞转录组学和单细胞蛋白质组学对 COVID-19患者血液中的免疫细胞进行分析,发现免疫细胞类型的组成发生改变。Specht团队[15] 利用开发的SCoPE2单细胞蛋白质组方法联合转录组学技术对巨噬细胞进行深入解析,在分选的 1 490 个单细胞中,鉴定到 3 042个蛋白质,发现巨噬细胞呈现明显的异质性。 Brunner 团队[53] 结合自己研发的单细胞处理平台与 diaPASEF 技术,分析了不同细胞周期各单细胞的蛋白表达差异,揭示了细胞周期的关键调控因子。目前针对哺乳动物体细胞的单细胞蛋白质组学在分析细胞间的差异蛋白、解析细胞间的异质性、以及发现新的细胞亚群方面,已经取得了一定的进展。未来随着技术的成熟,有望在生物医学研究中发挥更加重要的作用。
3 总结和展望
作为一个新兴的领域,随着质谱技术和样本制备技术的快速发展,单细胞蛋白质组学技术在近 5 年取得快速进展[10,37,39]。虽然许多学者已经开发出多种单细胞样品制备方法,但从获得生物样品到质谱检测,仍然存在肽段的吸附丢失等问题,单个哺乳动物细胞中只能鉴定到一千多种蛋白质,与单细胞转录组分析的深度相比,仍有较大的发展空间。单细胞蛋白质组学技术在样品前处理、色谱分离、质谱数据采集及数据分析等各个环节均可以进一步优化,减少样本损失,提高灵敏度,提升数据解析利用率。其次,单细胞蛋白质组学的通量较低。同位素标记技术大大提高了单细胞蛋白质组学分析的通量,但仍远远低于单细胞转录组学的测序通量。开发微流控、自动化装置等技术有望进一步提高单细胞蛋白质组学的分析通量,并减少人工操作可能带来的误差。此外,开发出更加适用于单细胞样品的专用色谱仪、质谱仪以及专门用于针对单细胞蛋白质组学质谱数据优化的算法,也将有力推动单细胞蛋白质组学的发展。
目前,单细胞转录组学与单细胞蛋白组学比较显示,单细胞蛋白组学结果比单细胞转录组学更稳定,两者具有很好的互补性[53];多组学联用技术将是未来科学研究的重要策略。此外,在蛋白质行使功能的过程中,蛋白质翻译后修饰发挥了重要作用,开发新的技术解析单个细胞内的蛋白质修饰信息,也是未来研究的一个重要方向。
随着各种相关方法的不断完善和成熟,单细胞蛋白质组学技术将不断提升,帮助我们从蛋白质水平解析细胞的调控与异质性,以及生理学和病理学中细胞异质性的分子基础;单细胞蛋白质组学技术有助于发现新的细胞亚群,对疾病进行分型分类,发现新的疾病亚型,指导临床疾病精准诊疗,均具有非常重要的意义。总之,单细胞蛋白质组学技术与单细胞转录组技术一样,将成为生物医学研究中的重要工具。
参考文献
[1]TIMP W,TIMP G.Beyond mass spectrometry,the next step in proteomics[J].Sci Adv,2020,6(2):8978-8993.
[2]AEBERSOLD R,MANN M.Mass-spectrometric exploration of proteome structure and function[J].Nature,2016,537(7620):347-355.
[3]WILKINS M.Proteomics data mining[J].Expert Rev Proteomics,2009,6(6):599-603.
[4]GEDDES-MCALISTER J,PRUDHOMME N,GUTIERREZ GONGORA D,et al.The emerging role of mass spectrometry-based proteomics in molecular pharming practices[J].Curr Opin Chem Biol,2022,68:102133.
[5]BANDURA D R,BARANOV V I,ORNATSKY O I,et al.Mass cytometry:technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry[J].Anal Chem,2009,81(16):6813-6822.
[6]BENDALL S C,SIMONDS E F,QIU P,et al.Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum[J].Science,2011,332(6030):687-696.
[7]CHALFIE M,TU Y,EUSKIRCHEN G,et al.Green fluorescent protein as a marker for gene expression[J].Science,1994,263(5148):802-805.
[8]CAPRIOLI R M,FARMER T B,GILE J.Molecular imaging of biological samples:localization of peptides and proteins using MALDI-TOF MS[J].Anal Chem,1997,69(23):4751-4760.
[9]LEVY E,SLAVOV N.Single cell protein analysis for systems biology[J].Essays Biochem,2018,62(4):595-605.
[10]ZHU Y,PIEHOWSKI P D,ZHAO R,et al.Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells[J].Nat Commun,2018,9(1):882-891.
[11]BUDNIK B,LEVY E,HARMANGE G,et al.SCoPEMS:mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation[J].Genome Biol,2018,19(1):161-172.
[12]CONG Y,MOTAMEDCHABOKI K,MISAL S A,et al.Ultrasensitive single-cell proteomics workflow identifies > 1000 protein groups per mammalian cell[J].Chem Sci,2020,12(3):1001-1006.
[13]DOU M,CLAIR G,TSAI C F,et al.High-throughput single cell proteomics enabled by multiplex isobaric labeling in a nanodroplet sample preparation platform[J].Anal Chem, 2019, 91(20): 13119-13127.
[14]TSAI C F,ZHAO R,WILLIAMS S M,et al.An improved boosting to amplify signal with isobaric labeling(iBASIL)strategy for precise quantitative single-cell proteomics[J].Mol Cell Proteomics, 2020,19(5):828-838.
[15]SPECHT H,EMMOTT E,PETELSKI A A,et al.Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2[J].Genome Biol,2021,22(1):50-76.
[16]DOU M,ZHU Y,LIYU A,et al.Nanowell-mediated two-dimensional liquid chromatography enables deep proteome profiling of < 1000 mammalian cells[J].Chem Sci,2018,9(34):6944-6951.
[17]WU R,XING S,BADV M,et al.Step-wise assessment and optimization of sample handling recovery yield for nanoproteomic analysis of 1000 mammalian cells[J].Anal Chem,2019,91(16):10395-10400.
[18]BERNHARD P,FEILEN T,ROGG M,et al.Proteome alterations during clonal isolation of established human pancreatic cancer cell lines[J].Cell Mol Life Sci,2022,79(11):561-582.
[19]BONNER W A,HULETT H R,SWEET R G,et al.Fluorescence activated cell sorting[J].Rev Sci Instrum,1972,43(3):404-409.
[20]YANG G,WITHERS S G.Ultrahigh-throughput FACSbased screening for directed enzyme evolution[J].Chembiochem,2009,10(17):2704-2715.
[21]VAN DEN BRINK S C,SAGE F,VERTESY A,et al.Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations[J].Nat Methods,2017,14(10):935-936.
[22]ESPINA V,WULFKUHLE J D,CALVERT V S,et al.Laser-capture microdissection[J].Nat Protoc,2006,1(2):586-603.
[23]WOO J,WILLIAMS S M,MARKILLIE L M,et al.High-throughput and high-efficiency sample preparation for single-cell proteomics using a nested nanowell chip [J].Nat Commun, 2021, 12(1): 6236-6246.
[24]DERKS J,LEDUC A,WALLMANN G,et al.Increasing the throughput of sensitive proteomics by plexDIA[J].Nat Biotechnol,2022,Online ahead of print.
[25]DAI X,SHEN L.Advances and trends in omics technology development[J].Front Med,2022,9:911861-911885.
[26]HUGHES C S,FOEHR S,GARFIELD D A,et al.Ultrasensitive proteome analysis using paramagnetic bead technology[J].Mol Syst Biol,2014,(10):757-770.
[27]SIELAFF M,KUHAREV J,BOHN T,et al.Evaluation of FASP,SP3,and iST protocols for proteomic sample preparation in the low microgram range[J].J Proteome Res,2017,16(11):4060-4072.
[28]ZHANG Y,YANG H,YU Y,et al.Application of nanomaterials in proteomics-driven precision medicine [J].Theranostics,2022,12(6):2674-2686.
[29]TSAI C F,ZHANG P,SCHOLTEN D,et al.Surfactant-assisted one-pot sample preparation for label-free single-cell proteomics[J].Commun Biol,2021,4(1):265-276.
[30]TURIAK L,OZOHANICS O,MARINO F,et al.Digestion protocol for small protein amounts for nanoHPLC-MS(MS)analysis[J].J Proteomics,2011,74(7):942-947.
[31]ZHANG P,GAFFREY M J,ZHU Y,et al.Carrier-assisted single-tube processing approach for targeted proteomics analysis of low numbers of mammalian cells [J].Anal Chem,2019,91(2):1441-1451.
[32]GUO Y,CAI L,LIU X,et al.Single-cell quantitative proteomic analysis of human oocyte maturation revealed high heterogeneity in in Vitro-matured oocytes[J].Mol Cell Proteomics,2022,21(8):100267-100282.
[33]TOTH G,SUGAR S,BALBISI M,et al.Optimized sample preparation and microscale separation methods for high-sensitivity analysis of hydrophilic peptides[J].Molecules,2022,27(19):6645-6651.
[34]LI S Q,SU K C,ZHUANG Z K,et al.A simple,rapid,and practical method for single-cell proteomics based on mass-adaptive coating of synthetic peptides[J].Sci Bull,2022,67(6):581-584.
[35]MASUDA T,INAMORI Y,FURUKAWA A,et al.Water droplet-in-oil digestion method for single-cell proteomics [J].Anal Chem,2022,94(29):10329-10336.
[36]XU K,LIANG Y,PIEHOWSKI P D,et al.Benchtop-compatible sample processing workflow for proteome profiling of < 100 mammalian cells[J].Anal Bioanal Chem,2019,411(19):4587-4596.
[37]LI Z Y,HUANG M,WANG X K,et al.Nanoliter-scale oil-air-droplet chip-based single cell proteomic analysis [J].Anal Chem,2018,90(8):5430-5438.
[38]PAN J Z,FAN C,ZUO Z Q,et al.Lab at home:a promising prospect for on-site chemical and biological analysis[J].Anal Bioanal Chem,2022,Online ahead of print.
[39]ZHU Y,CLAIR G,CHRISLER W B,et al.Proteomic analysis of single mammalian cells enabled by microfluidic nanodroplet sample preparation and ultrasensitive nanoLC-MS[J].Angew Chem Int Ed Engl,2018,57(38):12370-12374.
[40]GEBREYESUS S T,SIYAL A A,KITATA R B,et al.Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry[J].Nat Commun,2022,13(1):37-49.
[41]RUSSELL C L,HESLEGRAVE A,MITRA V,et al.Combined tissue and fluid proteomics with Tandem Mass Tags to identify low-abundance protein biomarkers of disease in peripheral body fluid:an alzheimer's disease case study[J].Rapid Commun Mass Spectrom,2017,31(2):153-159.
[42]CHEN X,SUN Y,ZHANG T,et al.Quantitative proteomics using isobaric labeling:a practical guide[J].Genomics Proteomics Bioinformatics,2021,19(5):689-706.
[43]CHEUNG T K,LEE C Y,BAYER F P,et al.Defining the carrier proteome limit for single-cell proteomics[J].Nat Methods,2021,18(1):76-83.
[44]SLAVOV N.Single-cell protein analysis by mass spectrometry[J].Curr Opin Chem Biol,2021,60:1-9.
[45]WILSON S R,OLSEN C,LUNDANES E.Nano liquid chromatography columns[J].Analyst,2019,144(24):7090-7104.
[46]GREGUS M,KOSTAS J C,RAY S,et al.Improved sensitivity of ultralow flow LC-MS-based proteomic profiling of limited samples using monolithic capillary columns and FAIMS technology[J]. Anal Chem,2020,92(21):14702-14712.
[47]SPECHT H,SLAVOV N.Transformative opportunities for single-cell proteomics[J].J Proteome Res,2018,17(8):2565-2571.
[48]CONG Y,LIANG Y,MOTAMEDCHABOKI K,et al.Improved single-cell proteome coverage using narrowbore packed nanoLC columns and ultrasensitive mass spectrometry[J].Anal Chem, 2020, 92(3): 2665-2671.
[49]XIE H,DING X.The intriguing landscape of single-cell protein analysis[J].Adv Sci,2022,9(12):2105932-2105950.
[50]ZHANG Z,HEBERT A S,WESTPHALL M S,et al.Production of over 27000 peptide and nearly 4400 protein identificationsbysingle-shotcapillary-zoneelectrophoresis-mass spectrometry via combination of a very-low-electroosmosis coated capillary,a third-generation electrokinetically-pumped sheath-flow nanospray interface,an orbitrap fusion lumos tribrid mass spectrometer, and an advanced-peak-determination algorithm[J].Anal Chem,2018,90(20):12090-12093.
[51]BEKKER-JENSEN D B,MARTINEZ-VAL A,STEIGERWALD S,et al.A compact quadrupole-orbitrap mass spectrometer with FAIMS interface improves proteome coverage in short LC gradients[J].Mol Cell Proteomics,2020,19(4):716-729.
[52]CINTRON-DIAZ Y L,GOMEZ-HERNANDEZ M E,VERHAERT M,et al.Spatially resolved neuropeptide characterization from neuropathological formalin-fixed,paraffin-embedded tissue sections by a combination of imaging MALDI FT-ICR mass spectrometry histochemistry and liquid extraction surface analysis-trapped ion mobility spectrometry-tandem mass spectrometry[J].J Am Soc Mass Spectrom,2022,33(4):681-687.
[53]BRUNNER A D,THIELERT M,VASILOPOULOU C,et al.Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation[J].Mol Syst Biol,2022,18(3):10798-10812.
[54]COX J.Prediction of peptide mass spectral libraries with machine learning[J].Nat Biotechnol,2022,Online ahead of print.
[55]MICHALSKI A,COX J,MANN M.More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS[J].J Proteome Res,2011,10(4):1785-1793.
[56]GUAN S,TAYLOR P P,HAN Z,et al.Data dependent-independent acquisition(DDIA)proteomics[J].J Proteome Res,2020,19(8):3230-3237.
[57]LIN L,ZHENG J,YU Q,et al.High throughput and accurate serum proteome profiling by integrated sample preparation technology and single-run data independent mass spectrometry analysis[J].J Proteomics,2018,174:9-16.
[58]HUFFMAN R G,CHEN A,SPECHT H,et al.DO-MS:data-driven optimization of mass spectrometry methods [J].J Proteome Res,2019,18(6):2493-2500.
[59]SCHOOF E M,FURTWANGLER B,URESIN N,et al.Quantitative single-cell proteomics as a tool to characterize cellular hierarchies[J].Nat Commun,2021,12(1):3341-3355.
[60]CHEN A T,FRANKS A,SLAVOV N.DART-ID increases single-cell proteome coverage[J].PLoS Comput Biol,2019,15(7):1007082-1007111.
[61]KALXDORF M,MULLER T,STEGLE O,et al.IceR improves proteome coverage and data completeness in global and single-cell proteomics[J].Nat Commun,2021,12(1):4787-4801.
[62]WANG B,WANG Y,CHEN Y,et al.DeepSCP:utilizing deep learning to boost single-cell proteome coverage[J].Brief Bioinform,2022,23(4):1-15.
[63]ZHANG Z,DUBIAK K M,SHISHKOVA E,et al.High-throughput,comprehensive single-cell proteomic analysis of xenopus laevis embryos at the 50-cell stage using a microplate-based MICROFASP system[J].Anal Chem,2022,94(7):3254-3259.
[64]SAHA-SHAH A,ESMAEILI M,SIDOLI S,et al.Single cell proteomics by data-independent acquisition to study embryonic asymmetry in xenopus laevis[J].Anal Chem,2019,91(14):8891-8899.
[65]VIRANT-KLUN I,LEICHT S,HUGHES C,et al.Identification of maturation-specific proteins by single-cell proteomics of human oocytes[J].Mol Cell Proteomics,2016,15(8):2616-2627.
[66]SCHULTE-SCHREPPING J,REUSCH N,PACLIK D,et al.Severe COVID-19 is marked by a dysregulated myeloid cell compartment[J].Cell,2020,182(6):1419-1440.
《生物医学转化》作者:霍子安,郭曰帅,王月,司徒成昊,郭雪江,生殖医学国家重点实验室,南京医科大学,江苏南京 211166;
文章编号:2096-8965(2022)04-0085-09
DOI:10.12287/j.issn.2096-8965.20220411