Aebersold研究组发布用于全蛋白质组的高通量定量方法
瑞士苏黎世联邦理工学院的研究人员领导的一个研究小组,已制定了一个质谱工作流,用于对整个蛋白质组进行高通量的绝对定量。
据ETH的研究者、该研究的领头人Ruedi Aebersold说,对于多种形态,该技术实现了目标蛋白质组的重现性定量,提供用于聚类分析(cluster analysis)的补充性数据,聚类分析被广泛地用于基因组工作(如转录组表征)中,但很少被用于蛋白质组学中。
发表在本周的分子系统生物学(Molecular Systems Biology)期 刊 上 的 论 文使用了该方法,科学家们在25种不同的细胞水平上,定量了1,680种人类病原体钩端螺旋体(pathogen Leptospira interrogans)的蛋白质,并获得了新的视角来洞察在组织的病原体发展和抗体防御方面蛋白质组的变化,
该工作代表了“第一次对于多种形态下的蛋白质组进行真正定量,已经完成。”Aebersold告诉记者说。“
“这使我们能将蛋白质组学研究移到一个和基因组社区(genomics community)相似的条件下,带有转录轮廓图,已经[能够]做十种、甚至是几十种形态,然后进行聚类分析。”从而使科学家们“在数据组之外获得信息,这些信息在单一的蛋白质中没有真正显现,但作为一个整体在蛋白质组的模式方面显现出来。”
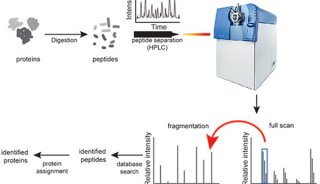
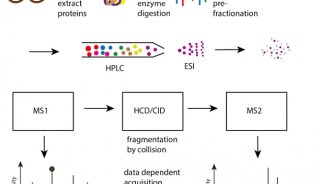
该技术使用一套LC-MS/MS来运行实验,以确定每个蛋白质最好的flying proteotypic peptides。它使用质量包含(mass inclusion)列表驱动的策略,把质量序列时间聚焦在这些最好的flying PTP上,将蛋白覆盖率最大化。对每个蛋白质三个响应最好的肽,关联它们信号强度的平均值和校正曲线,来建立一系列的同位素标记的参考多肽,最终获得待检蛋白质的定量。
该技术的定量“不是超精密的”Aebersold说道,估计它能检测到含量比≥1.5倍的差别蛋白,“但是,这种方法非常快”,比给每个蛋白质作重同位素肽标记,这种方法省钱多了。
Aebersold还说道,同样,该方法可对每个细胞中的蛋白拷贝数量进行绝对定量。
“今日在蛋白质组学中用到的更多的技术是相对定量,比如从一个点到下一个,蛋白含量增加了两倍或降低了两倍。”他说,“但是如果这种蛋白表达上的改变,一种情况是每个细胞从20到40个拷贝,一种情况是每个细胞从10,000到20,000个拷贝,两种情况的意义是不同的——比如可以估计能量消耗——所以这是附加的信息。
该方法在原则上相似于SRM,Aebersold说道,但它使用预估的参数进行质谱分析。为L. interrogans研究所需,测定这些参数要求做28次LC-MS/MS,或几天的质谱工作;当在不同的实验室、用不同型号的质谱测定时,这些参数证明可被转移。
相对于SRM-MS,该技术的最大优势是它的高通量,Aebersold说。SRM-MS实验典型受限于一次实验只能检测几百个目标肽段,而ETH的技术能在一次实验中检测成千上万个多肽。
该技术在动态范围方面受限。SRM-MS能够覆盖高达5个数量级的动态范围,新方法最多能够达到3或4个数量级。Aebersold说,“这使它最适合于研究诸如细菌这样的有机体,它们只有中等复杂度的蛋白质组。”
“两种技术想法一样”,他说,“你预先设定一些肽,然后您想具体分析。后来用两种技术得到不同的性能:一个是广泛但不深入,一个是深入但不广泛。”
他指出,一种新的叫做SWATH的质谱技术,能够同时具备新方法的深度和SRM-MS的广度。该方法建立在Aebersold实验室的AB SCIEX TripleTOF 5600质谱仪上,并在2011年ASMS年会上被AB SCIEX发布。
跟SRM-MS的采集一样,SWATH也是设特定的母离子窗口来获得目标碎片,期望该窗口中包括感兴趣的肽,并观测其碎裂离子的强度,从而对目标肽进行定性和定量。
差别是,Aebersold说,由于TripleTOF 5600质谱仪的速度特别快,SWATH可以选择一个25 amu的宽母离子窗口,并把所有的母离子质量范围划分成分段的25 amu宽的窗口,就可以“对液相流出的所有组分,获得所有的碎片”。
然后,使用Aebersold和系统生物研究所的Rob Moritz领导开发的SRMAtlas软件,研究者们就可以搜索、比对质谱采集的碎片离子谱图和SRMAtlas的参考谱库,从而鉴定多肽。
采用独特的搜索策略非常必要,Aebersold说,因为通过该技术产生的大碎片离子的谱图,会“呛(choke)”住一个典型的搜索引擎。
“因此我们使用SRM谱库,在这些组分谱里选择和发现特定的对目标肽响应的模式”,他说,“就像我们将用序列数据库搜索一样,我们不试图解释每张谱,我们只是想发现那些模式,能告诉我们一个特定的肽出现了,以及这个肽的含量。”
根据Aebersold的说法,最初使用该技术的研究显示,SWATH法能在复杂样本中鉴定超过4个数据量动态范围的蛋白,其定量的性能与SRM-MS相当。
“谈到检测的灵敏度,SWATH可能比SRM低3~4倍,但它接近于SRM的性能。”听说。
如果该方法实现了它的承诺,它可以提供“进一步的工具”,对一个蛋白质组的多种形态(如在MSB文章中所描述的那些形态)进行定量,Aebersold说,注意到“同样的心态下应用SWATH技术,可能进一步提高通量。”
相对于MSB技术,SWATH的一种局限是它在SRMAtlas上的可靠性,SRMAtlas目前只包括人、大鼠、酵母蛋白的参考谱库。另一方面,对于MSB方法来说,研究者们能够“快速流畅地生成参考谱库。” Aebersold说。
By Adam Bonislawski
A team led by researchers at the Swiss Federal Institute of Technology Zurich has devised a mass spec workflow enabling high-throughput absolute quantitation of entire proteomes.
The technique enables the reproducible quantification of a target proteome over multiple states, providing data amenable to the sort of cluster analysis that has been widely used in genomic work like transcriptome profiling but much less used in proteomics, said ETH researcher Ruedi Aebersold, leader of the study.
Using the method, which was described in a paper published this week in Molecular Systems Biology, the scientists quantified 1,680 proteins in the human pathogen Leptospira interrogans at 25 different cellular states, obtaining new insights into proteome changes involved in the organism's pathogen progression and antibiotic defense.
The work represents "the first time that really quantitative measurements on multiple states of a proteome have been done," Aebersold told ProteoMonitor. "This allows us to move proteomics to a similar situation as the genomics community, which with transcript profiling has [been able] to do profiles of tens or even dozens of states and then do cluster analysis," allowing scientists to "get information out of the dataset that's not really apparent from just a single protein but that is from the proteomic pattern as a whole."
The technique uses an initial set of LC-MS/MS runs to experimentally determine the best flying proteotypic peptides for each protein. This allows for a mass inclusion list-driven strategy that focuses MS-sequencing time on only these best flying PTPs, maximizing protein coverage. Quantification of the detected proteins is achieved by correlating the average of the signal intensities of the three best responding peptides per protein with a calibration curve built using a set of isotopically labeled reference peptides.
The quantification provided by the technique is "not super precise" Aebersold noted, estimating that it can detect differences of one-and-a-half-fold and greater, but, he said, "it is very fast" and vastly less expensive than adding heavy-labeled peptides for every protein.
Also key, Aebersold said, is that it provides absolute quantitation of(?) protein copy numbers per cell.
"Most techniques used in proteomics today give you relative quantitation, like from one point to the next a protein is up two-fold or down two-fold," he said. "But if [the expression changes] from 20 to 40 copies per cell, that has different implications than if it were to go from 10,000 to 20,000 copies per cell – for estimating the energy consumption, for instance – so this is additional information."
The method is in principle similar to selected-reaction monitoring, Aebersold said, in that it uses predetermined parameters to guide the mass spectrometer's analysis. Determining these parameters for the L. interrogans study required 28 LC-MS/MS runs, or several days of mass spec work, and these parameters proved transferrable when tried on a different model machine in a different laboratory, he noted.
The great advantage of the technique compared to SRM-MS is its throughput, Aebersold said. While SRM-MS is typically limited to several hundred targeted peptides in a run, the ETH technique can cover thousands of peptides in a run.
It suffers in terms of dynamic range, however. While SRM-MS can cover as many as five orders of dynamic range, the new method can go up to only three or four, making it best suited, Aebersold said, to studying organisms like bacteria that have only moderately complex proteomes.
"Both techniques use the same idea," he said. "You predetermine some peptides that you would like to specifically analyze, but then the two techniques have different performance profiles. One is broad but not as deep and the other is very deep but not as broad."
He noted that another new mass spec technique called SWATH could potentially offer both the breadth of the new method and the depth of SRM-MS. Developed by Aebersold's lab on AB Sciex's TripleTOF 5600 instrument, the method was introduced by AB Sciex at the American Society for Mass Spectrometry's annual meeting in June and has been described in a paper by Aebersold that is currently under review (PM 06/10/2011).
Like SRM-MS, SWATH targets for fragmentation particular precursor ion windows where it expects a peptide of interest to be present and then looks at the level of fragment ions to detect and quantify that peptide.
The difference, Aebersold said, is that, enabled by the speed of the 5600, SWATH selects a wide precursor window of 25 mass units and moves through the entire precursor ion mass range in segmented windows of 25 mass units, allowing it to "basically generate fragment ions from everything that is eluting from the [LC] column."
Then, using the SRMAtlas developed in an effort led by Aebersold and the Institute for Systems Biology's Rob Moritz, researchers search the fragment ion spectra captured by the machine against the reference spectra in the SRMAtlas to make the peptide IDs (PM 09/24/2010).
The unique search strategy is necessary, Aebersold said, because the large composite fragment ion spectra generated by the technique would "choke" a typical search engine.
"So we use the SRM libraries to select and find specific patterns in these composite spectra that correspond to target peptides," he said. "We don't try to explain every spectrum like we would with sequence database searching, we simply want to find patterns that tell us whether or not a particular peptide is present and in what quantity."
According to Aebersold, an initial study using the technique showed it could identify proteins in a complex sample over four orders of magnitude of dynamic range with quantitation equivalent to that provided by SRM-MS.
"In terms of sensitivity of detection, [SWATH is] probably a factor of three or four off from SRM, but it's approaching the performance of SRM," he said..
If the method fulfills this promise it could provide "a further tool" for quantitative measurements on multiple states of a proteome like those described in the MSB paper, Aebersold said, noting that "applying the SWATH technique with the same mindset would probably further increase throughput."
One limitation of SWATH compared to the MSB technique is its reliance on the SRMAtlas, which currently contains reference spectra only for human, mouse, and yeast proteins. For the MSB method, on the other hand, the researchers were able to "generate the reference library basically on the fly," Aebersold said.

 400-6699-117 转 8888
400-6699-117 转 8888
-
精英视角

-
市场商机

-
会议会展

-
综述
-
焦点事件

-
技术原理

-
焦点事件