400-6699-117转1000
咨询列表
北京阅微基因技术有限公司
您好,欢迎您查看分析测试百科网,请问有什么帮助您的?

诚信认证:
工商注册信息已核实! 扫一扫即可访问手机版展台
扫一扫即可访问手机版展台
| 参考报价: | 面议 | 型号: | 宏基因组测序 |
| 品牌: | 阅微基因 | 产地: | 北京 |
| 关注度: | 150 | 信息完整度: | |
| 样本: | 典型用户: | 暂无 |
400-6699-117转1000
宏基因组测序
宏基因组学(Metagenomics),又被称为微生物环境基因组学、元基因组学。其研究不要求对每种微生物进行分离、纯化和培养,而是以特定生物环境中总的微生物群落作为研究对象,提取全部DNA对其进行高通量测序、序列组装和基因注释,获得环境微生物基因信息的总和。以此可以分析微生物群体基因组成多样性与丰度、微生物的群落结构、物种分类、进化关系、微生物与宿主间的互作关系,和发掘新的或具备特定功能的基因等。宏基因组测序与传统的研究方法相比,具有实验周期短、通量高、速度快、信息全等优势,极大地扩展了微生物学的研究范围。
常见应用领域如下
医学领域:消化系统、内分泌系统、免疫感染类疾病,研究微生物与疾病关联等;
农业领域:植物根系微生物、施肥或农药影响土壤微生物、植物病害等;
畜牧领域:动物胃肠道、排泄物微生物与营养、疾病的关联等;
环境领域:土壤、水体、淤泥、雾霾,研究污染及生态修复、评价环境健康等。
特殊极端环境:矿井、深海、沙漠、火山口、温泉等极端环境条件下的微生物及资源研究。
技术路线

技术优势

测序策略
| 测序策略: | PE150 |
| 建议数据量: | 5-6G data |
收样要求
| 土壤 | 10 g |
| 粪便 | 2-5 g |
| 血液 | 10 mL |
| 污泥/沉积物 | 5-10 g |
| DNA | 总量≥ 500 ng,浓度≥ 20 ng/μL, OD A260/280:1.8 - 2.0, |
您可以得到的数据分析
生物信息分析流程

| 序列拼接与组装 | 物种注释及 群落结构分析 | 差异分析/ 关联分析 | 功能数据库 注释及分析 | 其他分析 |
| 基因预测 | 样品物种结构 Krona展示 | 主成分分析(PCA) | 基于KEGG、eggNOG、 CAZy数据库注释 | ★Network 网络分析 |
| 基因丰度分析 | MEGAN/GraPhlAn 可视化 | 基于物种丰度的 降维分析(NMDS) | 注释基因数目分析 | ★丰度共变化的基因 类群(CAG)分析 |
| 非冗余基因集 构建 | 注释基因数目及 相对丰度柱形图 | 主坐标分析 (PCoA) | 功能相对丰度聚类分析 | ★差异基因聚类 (MGS)分析 |
| Gene catalogue 基本信息统计 | 基于物种相对 丰度聚类分析 | 物种Anosim分析 | 基于功能丰度的降维分析 (PCA、PCoA、NMDS) | |
| Core-pan 基因分析 | 组间差异物种的 LDA/LEfSe分析 | 基于功能丰度的 Anosim分析 | ||
基因数目差异分析 (组间基因数目差异 箱图,Venn图) | 物种MRPP分析 | ★基于ARDB数据库 抗生素抗性基因(ARGs) 注释及相关分析 | ||
基因数目的样本间 相关性分析 | Welch's t-test分析 | |||
★环境因子关联分析 (CCA/RDA分析) |
注:★代表高级分析
数据分析图例


微生物组学与代谢组学关联分析图例
1、微生物与代谢产物关联分析热图

2、微生物与代谢产物(大类)关联分析热图

3、微生物与代谢产物关联网络图

4、典型微生物/代谢产物散点图分析

5、差异代谢通路与功能的分析

6、差异菌种代谢功能与代谢产物关联分析

案例解析
牛瘤胃宏基因组测序绘制出913个微生物基因组图谱
瘤胃作为牛的消化器官,能将植物材料分解并转化为能量,这主要由瘤胃中微生物的协同作用完成,科学家希望通过了解这个过程来改善牛的消耗—产出比。目前在公共数据库中对牛瘤胃菌种信息和其对应功能的收录并不完整,来自英国爱丁堡大学的研究人员使用宏基因组测序和基于Hi-C的proximity-guided组装,从超过800 Gb的牛瘤胃的宏基因组测序数据中组装得到913个细菌和古菌的基因组草图。
研究者利用宏基因组测序分析42头苏格兰牛的瘤胃微生物群,得到共768Gb的数据,组装出 850个RUGs(rumen uncultured genomes),接着对第43个样品进行宏基因组测序结合Hi-C技术辅助组装,产生了额外的63个基因组草图(完整性≥80%,污染率≤10%),称为hRUGs(Hi-C rumen uncultured genomes)。
物种注释的结果有7个RUGs可注释到种,158个注释到属,而至少46%可注释到科。在可解析到门水平的895个RUGs中,厚壁菌占优势(50%),其次是拟杆菌(36%),放线菌(3.5%),变形菌(3.1%),广古菌(3.1%)和螺旋体(1%),代表了牛瘤胃中发现的主要微生物类型。该研究中得到的913个组装基因组含有1,979,391个预测的蛋白序列,在CAZy数据库中进行比对和过滤,总共有69,678个序列比对到至少一个碳水化合物酶活性功能,而其中只有6,061个(8.7%)在多个已知蛋白数据库中有高度相似的匹配(≥95%的一致性),这表明研究者通过组装基因组预测到很多新的蛋白。该研究通过高深度的宏基因组测序技术,并联合Hi-C得到的913组装基因组将会改变我们对瘤胃宏基因组学数据的解读方式,具有重要的研究价值。
Stewart et al., “Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen”,Nature Communications(2018) DOI:10.1038/s41467-018-03317-6

A. 913个基因组草图绘制的系统发生树,图中可见几个大分支。两个优势菌属分别为梭菌目和拟杆菌目,其中后者的一个重要簇代表为普氏菌科(Prevotellaceae)。较小的进化枝代表变形杆菌、古细菌、放线菌、螺旋体和纤维杆菌。其余节点和分支代表杂菌。
B. 物种丰度聚类热图,横坐标为样本,纵坐标为菌种的丰度。
宏基因组测序揭示强直性脊柱炎病人特异的肠道微生物标记物
强直性脊柱炎(Ankylosing Spondylitis,AS)是一种慢性炎性疾病,严重危害人们的身心健康和生活质量。AS在汉族中的发病率为0.2-0.54%,有8-10年的潜伏期,早期症状不易诊断,且病因尚不明确。先前认为AS与遗传密切相关,但近期科学家证明肠道微生物在AS的发病过程中起到重要的作用。本研究利用宏基因组测序技术研究AS患者与健康人的肠道菌群,旨在揭示肠道微生物在AS发病过程中可能的分子机制,以及通过肠道菌群作为潜在可利用的微生物标记物进行AS疾病的早期发现和鉴别。
本研究采用73个AS病人和83个健康人的探索队列分析比较了不同分类水平上两组的肠道微生物组成差异。与健康组相比,AS患者肠道菌整体物种丰度降低,梭杆菌门和疣微菌门丰度降低,肠杆菌属和柠檬酸杆菌属等革兰氏阴性菌几乎消失,而放线菌门细菌丰度显著升高。接着,构建AS患者的基因集,与健康人比较,患者肠道菌的数量和基因表达量都下降。测序结果显示有23,709个基因在组间存在表达差异。和二型糖尿病、慢性肝炎疾病肠道菌相似,AS患者的肠道菌基因集富集到membrane transport及metabolism of cofactors and vitamins,而在cell motility、signal transduction基因通路等则较健康人表达量降低。通过对患者差异表达基因的关联分析和聚类筛选,三类生物标志被挑选建模,分别为33个参照基因组(物种)、30个基因和62个基因簇。
随后将三种AS诊断模型以24个AS病人和31个健康人队列进行验证评价,诊断率(鉴别率)均较高,其中又以基因标志效果最佳(AUC=96.64%)。本研究通过肠道微生物宏基因测序初步建立AS疾病的诊断模型,并推测肠道菌群在AS炎症发生过程中可能的分子机制,对AS疾病的诊断和治疗有着重要意义。
Wen et al., “Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis”Genome Biology(2017) DOI 10.1186/s13059-017-1271-6
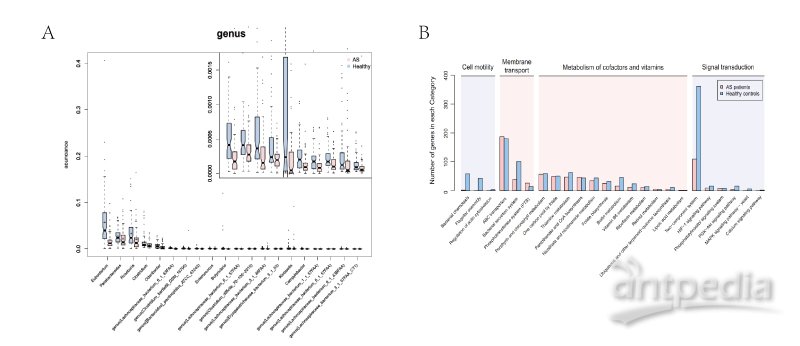
A. 宏基因组测序后,健康人与AS病人在种水平上的菌株差异箱线图。粉色代表AS病人,蓝色代表健康人。
B. 健康人与AS病人的四类KEGG代谢通路直系同源组(KEGG orthology)的基因数,粉色代表AS病人,蓝色代表健康人。
研究趋势与研究热点
宏基因组研究是对微生物群落总基因进行高深度测序,在鉴定微生物群落的同时,主要注重挖掘功能基因及其代谢途径。基于测序技术和生物信息学的快速发展,宏基因组技术优势在微生物领域中愈发明显,应用范围也愈发广泛。
应用方向:
1.构建特定环境下的完整微生物谱
2.寻找特定疾病或亚健康人群的菌群标志物
3.特殊微生物群体致病机制研究
4.耐药性和抗性基因筛查
5.微生物潜在基因资源挖掘
客户常见问题
扩增子测序和宏基因组测序的区别是什么?
两者都是评估特定生境总体微生物组成的高通量测序方法,都无需分离培养菌群。16S rDNA测序指对样本的总体微生物群落的16S rDNA 可变区(V1-V9区)的特定一段进行PCR扩增和测序。对细菌和古菌物种组成和丰度进行有效的鉴定。宏基因组测序是直接对片段化后样本的总体DNA进行测序,得到生境中全部微生物遗传物质的总和, 并通过大数据量的统计组装得到更深度的信息(基因组序列、基因信息、基因功能、微生物群落功能、群落中各成员之间的代谢网络等)。过程中无PCR扩增,避免误差,结果更忠于样品的真实性。
对于宏基因组项目,如何排除宿主污染?
肠道微生物的取样时,难免会遇到宿主污染的问题,遵循以下建议可以减少宿主污染。然而一旦发生较大的宿主污染情况,则需要利用已知的宿主基因组序列在数据分析流程中去污染。
(1)取样时,尽量不要取靠近组织的部位;
(2)提取的时候采用相应的试剂盒;
(3)若有参考基因组,可通过比对分析去除宿主基因组污染。
知名合作伙伴
企业资质
服务部分被引用文献(201908 部分 IF 大于5)
宏基因组测序信息由北京阅微基因技术有限公司为您提供,如您想了解更多关于宏基因组测序报价、型号、参数等信息,欢迎来电或留言咨询。
注:该产品未在中华人民共和国食品药品监督管理部门申请医疗器械注册和备案,不可用于临床诊断或治疗等相关用途