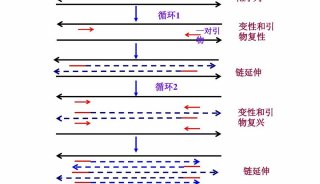
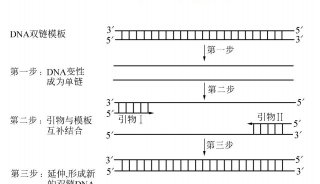
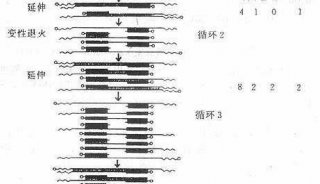
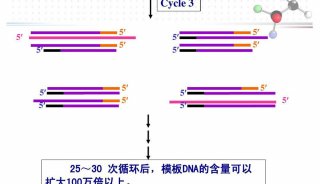
PCR污染的处理(三)
(二)反应液污染
可采用下列方法之一处理:
1. DNase I法:PCR混合液(未加模板和Taq聚合酶)加入0.5U DNase I,室温反应30 min后加热灭活,然后加入模板和Taq聚合酶进行正常PCR扩增。该方法的优点是不需要知道污染DNA的序列;
2. 内切酶法:选择识别4个碱基的内切酶(如Msp I和Taq I等),可同时选择几种,以克服用一种酶只能识别特定序列的缺陷,室温作用1h 后加热灭活进行PCR;
3. 紫外照射法:未加模板和Taq聚合酶的PCR混合液进行紫外照射,注意事项与方法同上述UV照射法;
4. g射线辐射法:1.5kGy的辐射可完全破坏0.1ng基因组DNA,2.0 kGy可破坏104拷贝的质粒分子,4.0 kGy仍不影响PCR,但高于此限度会使PCR扩增效率下降。引物可受照射而不影响PCR,g射线是通过水的离子化产生自由基来破坏DNA的。
(三)尿嘧啶糖苷酶(UNG)法
由于UV照射的去污染作用对500bp以下的片段效果不好,而临床用于检测的PCR扩增片段通常为300bp左右,因此UNG的预防作用日益受到重视和肯定。
1. 原理:在PCR产物或引物中用dU 代替dT。这种dU化的PCR产物与UNG一起孵育,因UDG可裂解尿嘧啶碱基和糖磷酸骨架间的N-糖基键,可除去dU而阻止TaqDNA聚合酶的延伸,从而失去被再扩增的能力。UNG对不含dU的模板无任何影响。UNG可从单或双链DNA中消除尿嘧啶,而对RNA中的尿嘧啶和单一尿嘧啶分子则无任何作用。
2. dUTP法:用dUTP代替dTTP,使产物中掺入大量dU。在再次进行PCR扩增前,用UNG处理PCR混合液即可消除PCR产物的残留污染。由于UNG在PCR循环中的变性一步便可被灭活,因此不会影响含dU的新的PCR产物。
3. dU引物法:合成引物时以dU 代dT,这样PCR产物中仅5ˊ端带dU。UNG处理后,引物失去了结合位点而不能扩增。对长片段(1-2kb以上)的扩增用dUTP法效率较用dTTP低,而用dU法就可克服这一缺点。dU引物最好将dU设计在3ˊ端或近ˊ端。该法仅能用于引物以外试剂的处理。
4. 优点:可以去除任何来源的污染;UNG处理可以和PCR扩增在同一个反应管内进行;由于扩增产物中有大量dU存在,可彻底消除污染源。
5. 需注意的是掺入dUTP的DNA不应对产物的任何操作有影响,在进行PCR产物克隆时,应该转化UNG-(UNG缺陷)大肠杆菌受体菌,否则转化产物会被受体菌UNG消化掉。
(四) 固相捕获法
用于去除标本中污染的核酸和杂质,原理如下:1)用一生物素标记的单链RNA探针与待扩核酸杂交,杂交区域是非扩增区;2)用包被链霉亲和素的固相载体来捕获带有生物素探针的杂交核酸,通过漂洗可去除污染的扩增产物和杂质;3)洗脱靶分子后用特异引物扩增非RNA探针杂交区域。第2)步的漂洗后可用PCR检测以确定标本是否被扩增产物或重组质粒污染。
(五)RS-PCR法(RNA-specific PCR)
也称为链特异性PCR,主要指用于RNA模板的特异性PCR法,该法可明显降低假阳性而不影响PCR的敏感性。其关键在于设计引物,逆转录引物的3ˊ端(A区)有2 0个核苷酸左右为模板的特异性互不序列,5ˊ端2 0个核苷酸(C区)为附加修饰碱基。与mRNA逆转录后,经超速离心使 cDNA与多余引物分开,再用和第二引物(C)以第一链cDNA为模板合成第二链cDNA,以后的PCR循环中用逆转录引物的B区和引物C进行扩增加尾cDNA,而污染的DNA或质粒DNA才不会被扩增。
(六)抗污染引物法
该对引物扩增时通过病毒DNA克隆如入质粒的位点。这一区域只存在完整的原病毒中,在重组质粒中,这一区域分成两个区域与克隆位点被。如果重组质粒污染了标本,也不能扩增出任何条带,即使出现了扩增带,其大小也与预期的不同。只有原病毒DNA才能被引物扩增,因此只要出现预期大小的扩增带就可以证明标本是阳性的,该法试用于环状靶分子系列。
四.其它PCR检测方法:
(一) 两步法:是指在用套式PCR方法扩增某些含量低微的标本时,两对引物在同一个管中以简化步骤,减少污染。通常第一步进行20-25个循环,扩增外引物片段;第二步再进行10个循环,扩增内引物片段。两步法对内外引物的Tm值有特殊要求,即内外引物的退火温度高(如68℃),在此温度下内引物不与模板退火,然后降低退火温度(至55℃)再扩增内引物片段,这样,在操作过程中,仅打开PCR反应管一次,大大减少了污染的可能性。
(二) 荧光法:亦称荧光PCR技术(fluoresence PCR, F-PCR),是1995年由美国PE公司首先研制成功的,它融汇了PCR的灵敏性、DNA杂交的特异性和光谱技术精确定量的优点,电脑同步跟踪,数据自动化处理,直接探测PCR过程中的变化以获得定量的结果,不需要做PCR后处理或检测,完全闭管操作。探针标记除用TET和FAM外,还可用HEX、JOE作为报告荧光,3′端的淬灭基团常用TAMRA。在探针保持完整时,荧光报告基团的荧光被荧光抑制基团淬灭,而在探针被切断后,荧光报告基团才发出报告荧光,且荧光的强度与PCR产物的数量呈正比。荧光检测仪器透过PCR管壁能直接检测到荧光信号的波长和长度变化。
主要有以下优点:
1.探针特异性强,假阳性率低;
2.操作快速,不需要PCR后处理;
3.定量范围宽,HBV 0.4fg-4000fg/ml(102-106 Dane′s particle/ml);
4.闭管操作,PCR产物污染少;
5.灵敏度高。
主要方法有:
1..荧光探针法:进行荧光PCR检测时,要求荧光探针必须完全与靶基因互补,长度以20-30个碱基为宜,必要时3′端磷酸化封闭,以防在扩增时作为引物延伸。该方法利用Taq酶的3′→5′聚合酶活性及5′→3′外切酶活性,可以在链延伸过程中实现链替换,并将被替换的探针切断,故可进行定性与定量检测。
荧光探针5′端标记的TAMRA,在480nm激发下产生530nm的红色荧光;3′端标记的FAM,激发后产生绿色荧光。PE公司推出的TaqMan系统,即采用双荧光探针,如配合使用PCR扩增与荧光检测合二为一的仪器,可进行实时(real-time)定量检测。
2.分子信标法:分子信标(molecular beacon)是一个发夹样结构的特异探针,其环状部分与靶序列互补。在室温时,分子信标的发夹紧闭,荧光淬灭。PCR扩增时,随着温度升高,发夹松开,与单链模板特异结合,发出荧光。荧光强度与模板呈正比,故可用于PCR产物的定性及定量。
总之,人类在迈入廿一世纪中即将出现若干的突破,生物医学便是其中重要的一项。在过去三、四十年耒,像PCR这样影响深远的技术实在很难找到。它的震撼, 除了众多的得奖外(包括诺贝尔奖),更在于它的可塑性、修饰性及全方位的运用。未来的生物医学领域中,它也必定继续扮演举足轻重的角色。