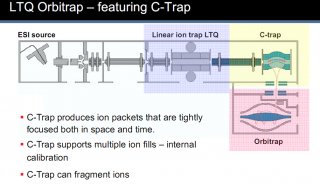
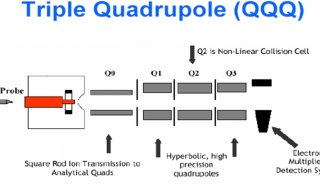
John Yates专访:探索质谱与信息学之间的奇妙关系
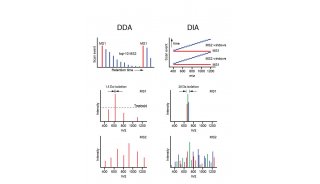
计算机对质谱的发展一直起着巨大的影响。从数据采集到仪器操作再到数据分析,计算机在质谱发展史中的多个关键时刻都起到了积极推动作用。串联质谱和信息学的结合,使蛋白质组学能够快速将氨基酸序列与质谱图进行归属。
随着分析工具的日渐发展、数据生产数量与数据类型的逐渐多样化,计算机信息的处理能力也在不断增长以与数据发展相适应。人们正在将高级的工作流程用于处理蛋白质组实验,包括数据的搜索、定量和统计分析。如今质谱仪又增加了离子淌度这一新的维度,这为数据采集、肽段的信息注释提供了更强的分析能力。IP2是一个蛋白质组学平台,它创建了一个结合GPU搜索、灵活定量和数据统计分析的工作流程。
Technology Networks近期与John Yate教授和Robin Park博士进行了访谈,以了解有关他们所开发软件IP2的更多信息,及其在蛋白质组学研究中的作用。(https:/www.technologynetworks.com/)

Q:请告诉我们质谱与计算机之间不断发展的关系?
多年来,质谱分析和计算机存在着一个有趣的关系。有一个概念被称为“邻近可能”(由Stuart Kauffman在2002年提出)。他指出,在任何限定的相邻时间范围内,一起协作的事物都会逐步的发生演化并产生创新。这个想法与质谱发展极为相似。若回顾质谱的发展历史,我们会发现,许多质谱的早期工作也是正在开发计算机的学术机构中进行的。
这促使了人们逐渐在各种研究项目中使用计算机,例如计算复杂而精确的分子质量。 最终,技术的发展让人们开始使用计算机记录质量,淘汰了传统的相机底片。随着时间的推移,计算机程序开始更高效地处理数据,让人们从中获得了更多的有效信息。
Q:众包和当前技术在质谱学发展中有多重要?
非常重要。在计算机技术发展的同时,人们意识到需要避免重复解释质谱图。这促使人们诞生了创建谱图库的想法,这些谱图库中的信息是已经被人们注释过的,而这种做法实际上开启了图库搜索的概念。这是科学界“众包”已知的早期例子。
在当时,计算机并没有装配大量的存储空间或内存,因此搜索程序必须设计的非常智能,以减少搜索谱图库所需的信息和技术压力。计算机控制的数据采集被依赖数据采集所替代,然后是独立数据采集,最终允许计算机对肽段进行大规模分析。现在人们可以处理肽和氨基酸序列条形码的串联质谱图,并用它来扫描数据库并识别所代表的氨基酸序列。
这种新的分析方法让高通量和大规模实验更能适应高度复杂的生物系统,包括蛋白质复合物、细胞器、细胞和相关组织。同时,这会产生更多的数据和并需要更多的分析,需要人们有更充分的准备。
Q:信息学的发展如何影响质谱的发展潜力?
质谱技术的发展给我们收集、分析、组织和解释数据的能力带来了很大压力。这就使信息学变得尤为重要,从数据提取、搜索引擎、定量分析到验证工具,置信度评分工具和数据存储库变得司空见惯——后者经常使用Microsoft Excel作为存储介质。然而它并不理想,所以一直以来,我们在逐步放弃使用。
能够在实验室中使用数据对我们来说非常重要,因此和布鲁克合作,我们一直在开发蛋白质结构分析和基因富集分析工具。
Q:您能介绍一下IP2平台吗?
几年前,我们在多款数据提取工具之间进行了比较,发现它们的功能存在很大的差异。 这是流程中最重要的步骤之一,但是拥有这么多软件工具的问题在于,很难在整个工作流程中实现完美的兼容性。为此,我们成立了一家名为Integrated Proteomics Applications的公司,开发了一个名为Integrated Proteomics Pipeline(IP2)的工作流框架。这个框架背后的想法是,我们可以将人们在学术界开发的工具(免费且开源的工具)放入到这个IP2框架中,创建了一个集成数据分析工具的简化工作流程。
IP2是一个中间层程序,它用于管理分析、谱图质量控制、后端层、到链接云和集群计算、数据存储和备份。用户可以使用计算机、手机或平板电脑访问他们的数据或查看进程状态。 IP2也可以通过开发人员工具包进行设置,这意味着用户可以调整IP2平台以与其它软件和应用程序协同工作。
IP2可以用于大规模并行蛋白质组学数据分析,我们已经能够通过亚马逊网路服务系统、谷歌和微软云系统将我们的平台与云计算结合。我们还将我们的实验室系统和云系统,通过高性能GPU集群来提高平台的分析速度和效率。

Q:GPU如何影响IP2的性能?
IP2允许使用GPU搜索引擎而不是CPU搜索引擎。相较之下GPU速度更快。一个GPU卡上拥有数千个GPU核心,用户可以添加更多的GPU卡来提高速度。GPU搜索数据库的速度惊人,并且GPU的数据库搜索能力也会随计算能力增长而提高。

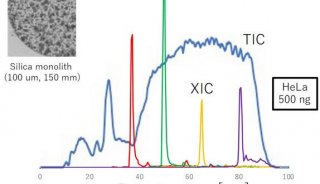
图:基于GPU计算的IP2将数据处理速度提升百倍
我们一直在考虑使用双重搜索来提升数据质量,并希望将此功能整合到IP2中。在这里,您将搜索到的DDA数据传递至序列数据库,然后使用该数据构建相关数据库。再使用该库再次搜索DDA数据。这种方法可以显著提高数据的再现性,GPU内核的使用即为我们实现了此目标。
Q:timsTOF质谱平台如何与IP2集成?
布鲁克的timsTOF是测量离子淌度的极佳工具。这类质谱可为我们提供有关离子三维结构的信息,有助于提高峰容量和化合物表征的可靠性。我们一直在为timsTOF优化我们的平台、工具和搜索引擎,特别是围绕如何从timsTOF的原始文件中提取数据的过程这个问题,这些原始文件信息量着实不小。
通过这项工作,我们建立了一个强大的提取程序:timsTOF Extractor,ProLuCID搜索引擎(使用我们之前提到的GPU技术),Census定量分析应用程序和用于timsTOF的PaSER(并行实时数据库搜索引擎)。
Q:在开发宏蛋白质组学和微生物组数据分析搜索引擎工时有哪些挑战?
微生物组数据库庞大,处理微生物数据具有极大的挑战性。目前它的大小约为70G,并且还在不断增长。在传统的搜索策略中,使用这种方法搜索大数据是很困难的。为了解决这个问题,我们与Scripps研究所的Dennis Wallen合作开发了ProLuCID-ComPIL搜索引擎。
ProLuCID-ComPIL引擎使用NoSQL对数据进行预分类和预分析,以缩短用户搜索时间。 这些算法和搜索过程也可以与PTM和序列变量一起使用,把它们传输到索引数据库中,然后使用GPU对其进行高速搜索。我们与加州大学圣地亚哥分校的Yu Gao亲密合作,使用他的谱图校准工具Dilu,我们可以专心把注意力放在代谢组学上。
Q:您能告诉我们更多有关PaSER系统的信息吗?
我们的PaSER系统是一个并行数据库搜索引擎,可以实时工作。由于许多仪器配套软件扫描速度非常快,生成大量谱图,因此实时搜索的一个关键优势是省略了数据提取步骤--你只需直接从质谱仪中获取数据,然后立即搜索,无需再上传数据。
我们研发的PaSER系统速度足够快,可以同时接受多个仪器数据,当然了,我们使用PaSER平台的目标不仅仅是实时搜索。希望能继续解决实时搜索功能方面存在的更多挑战。传入搜索引擎的仪器扫描速度要想提高,搜索引擎的速度至关重要。与IP2平台一样,PaSER使用GPU内核而不是CPU内核,比传统搜索速度更快。这意味着可以将数据从仪器实时发送到IP2-GPU中心,并且在实验完成后立即获得数据库搜索结果。
Q:与传统的离线搜索相比,如何评价PaSER在速度方面的提升?
为了评估PaSER的性能,我们在timsTOF Pro上运行了200纳克的HeLa细胞样品。整个过程我们重复进行了六次。第一次实验是使用实时搜索,第二到第四次没有使用,在第五和第六次实验中使用了两次实时搜索。这项实验的目的是识别扫描速度是否滞后,并检查使用实时搜索是否会影响成功识别的次数。
通过实验我们发现,使用实时搜索不会影响扫描性能。最后,我们还发现使用实时搜索能获得与离线搜索相同数量的结果。
此外,我们还评估了离线搜索所花费的时间。有时,在使用实时搜索之后,可能会再次使用不同的参数在不同的数据库中继续搜索。这种情况下,用户并不需要转换原始数据,因为最初的实时搜索已经保存谱图信息并将其传输到数据库。在我们的演示中,每次使用IP2-GPU引擎搜索,只需花费3分钟。

Q:Smart Precursor Selection工具是如何改善搜索过程的?
PaSER的智能预选工具能够实现PaSER与仪器间的双向联系。根据不同目标,能实现更多的创新。例如,我们可以使用此工具来实现排除列表。 这个想法很早就有了,但是由于并不容易实现,所以并不受广大用户欢迎。过去,用户必须在搜索结果中手动收集肽段ID,再手动输入到排除列表。随着项目的发展,人们依然将重复这个复杂的过程。
现在,PaSER可以从第一个实验开始就自动建立排除列表,然后将此表传递给第二个实验,依此类推,每次的迭代都会完善排除列表。这种方法使我们的搜索和实验随着时间推移产生更准确的结果。
另一个是仪器环境温度变化或校准问题而引起的质量漂移动态管理。通过使用PaSER的实时搜索功能,我们可以测量理论和实际肽的母离子间质量差。然后,通过将增量质量发送回仪器中,实现实时校准离子质量漂移。这意味着无论我们完成十次运行还是一百次运行,质量校准始终是最新的。
当我们使用基于MS1的定量分析进行标记时,我们经常会看到同一肽的轻离子和重离子。对于定量分析,因为我们已经知道轻离子和重离子之间的质量差,所以不需要同时使用两种前体离子,一个就足以定量样品。
在实时搜索过程中,我们就能确定到底是在处理重肽还是轻肽,然后我们可以动态排除另外一对,也不会因此生成多余的谱图。
我们还可以使用这项工具来处理工作站中的特定标签,例如AHA标签或TEV标签。 其中,AHA标签可以标记蛋氨酸,而TEV标签可以标记半胱氨酸。根据UniProt数据库计算,大约65%的肽不含半胱氨酸或蛋氨酸,利用这些数据,我们可以实时排除许多肽段,并根据需要进行选择性扫描。

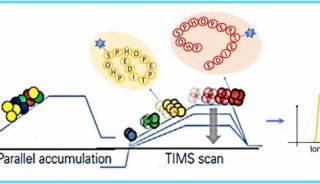
Q:PaSER如何适应动态PASEF谱图和实时定量?
PaSER可以使用PASEF技术或并行累加串行分段作为其操作的一部分。在每个PASEF循环中,我们能通过构架来构建PASEF扫描。若根据离子丰度,一个PASEF循环可能会因缺少足量的离子导致无法获得结果。而我们的实时搜索功能可以实时评估谱图。如果引擎在检查中发现需要扫描更多信号时,则可以通过添加更多构架来加强信号。
以前,我们需要知道峰值才能有效触发串联质谱图,现在可以在实时搜索过程中通过评估谱图来确定峰值后,仅触发母离子实现这一点。我们也可以在最合适的时段从排除列表中动态删除无用的离子。
最后,我们利用timTOF开发实时定量功能。该功能通过离子淌度来计算肽的量,可以代替以往的XYZ峰面积。我们还可以从实时搜索中分离具有ID的共洗脱肽/肽段,这意味着人们可以实现并行定量分析。如果在进行多个实验时,我们可以在实验间建立匹配关联。这些都是PaSER双向联系功能创造性使用的优秀范例。
Q:IP2和布鲁克的其他平台在更广泛的数据和知识行业中处于什么样的位置?
现在,我们拥有各种各样的工具,可以对质谱数据进行分类和识别 。当然,我们的总体目标是更好的进行生物探索。考虑到这一点,我们将应用程序链接到诸如Reactome等数据分析工具,其中有许多工具可以在互联网中免费使用。有一些工具能探索基因本体论的输出,例如,通过选择最重要的20个类别,同时在生成的原始文件中保留所有类别的信息,接下来可以直接进行下一步检查而无需重新分析。
Mathieu Lavallée-Adam和他的团队为我们开发了一种内部工具——PSEA-Quant,专门用于蛋白质集富集分析,是专为基因集富集分析而开发的,但已针对无标记和基于标记的蛋白质定量数据进行了优化。
总体而言,协调分析平台和研究工具之间的兼容性是我们工作的重点。