| 实验步骤 | 一、第一向
1. 固相 pH 梯度凝胶或凝胶条的准备
固相 pH 梯度凝胶的灌注及聚合方法请参阅 固相 pH 梯度等电聚焦 有关章节,如需要可切成 3~5 mm 宽的胶条在 -20℃ 保存一年。为避免繁琐和复杂的灌胶和切胶条程序和保证重复性,可从市场上购置凝胶条使用。市场上有不同长度和不同 pH 范围的固相 pH 梯度凝胶干胶条可供选择,长度最短为 7 cm,最长为 24 cm。pH 范围有宽范围,窄范围和极窄范围。GE 医疗集团、安玛西亚公司在 pH 范围方面有更多的选择,见图 8.4。凝胶条长度的选择取决于第二向水平或垂直电泳槽的宽度,pH 范围的选择则取决于分辨率的要求和样品的特性。

在一些 pH 范围,凝胶条分为线性和非线性(non-linear, NL) pH 梯度两种。前者是指 pH 梯度在凝胶中线性分布。后者是指在凝胶中的某一区间 pH 梯度的斜率小,因而在此区间的分辨率高,如图 8.5 (b) pH 3~10 NL 中 pH 5~7 的三个斑点(椭圆中)比图 8.5 (a) 的三个斑点要分得开。

2. IPG 凝胶干胶条的再水化(泡胀)
为了样品等电聚焦的需要,作为双向电泳第一向的固相 pH 梯度凝胶条中必须含尿素,去污剂,还原剂,载体两性电解质等,不能长期保存,故按 固相 pH 梯度等电聚焦 制成的(或市售的固相 pH 梯度凝胶)干胶条必须用含有这些成分的水化溶液重新泡胀。
将在 -20℃ 保存的 IPG 干胶条在室温平衡一段时间后,去掉保护膜。用新鲜配制的泡胀液重新水化凝胶。为保证凝胶的厚度和浓度,应将干胶条放在模具中泡胀过夜。
IPG 凝胶干胶条可以在室温泡胀,也可以在电场(30V,20℃ ) 中泡胀。泡胀液中可以含样品,也可以不含样品,对不稳定样品应采用后一种。泡胀至少需要 10 小时以上,建议在晚上进行。为防止泡胀液蒸发和尿素结晶,要在 IPG 凝胶干胶条上铺一层覆盖液(通常用硅油 )。具体操作方法请参阅仪器说明书。
泡胀液的组成取决于蛋白的溶解需要。通常含有尿素、非离子或两性离子去污剂、还原剂,有时也含有 IPG 缓冲液(通常在含有样品时泡胀应加 IPG 缓冲液 )。泡胀液的配制参见表8.2。

含有 IPG 缓冲液的泡胀液是在使用前按表 8.2 所述的泡胀液中加 IPG 缓冲液,终浓度为 0.5%~2%,所用 IPG 缓冲液的 pH 范围与凝胶干胶条的 pH 范围相同或尽可能相近。载体两性电解质有时也可以代替 IPG 缓冲液。
3. 样品制备
样品制备是影响双向电泳结果的主要因素之一。为了提高分析复杂蛋白混合物的分辨率,首先必须使感兴趣的蛋白得到充分的溶解。对可溶蛋白的样品,如血清,体液只需去除白蛋白和 IgG,再作简单处理便可直接用于双向电泳。对细胞,特别是组织,一般都需经过破碎、裂解、沉淀、溶解等多次处理才能上样。裂解液和样品液的组成见表 8.3。

样品液与裂解液相似,但无需再加蛋白酶抑制剂。如需观察,可加少许溴酚蓝。通常要用 4 倍的样品液溶解裂解后的样品。如果样品中有较多的盐,则应用更高比例的样品液溶解。对疏水蛋白等较难溶的蛋白可用 5 mol/L ( 或 7 mol/L ) 尿素,2 mol/L 硫脲,4% CHAPS,60 mol/L DTT,2% IPG 缓冲液(或载体两性电解质 )。
2004 年在北京召开的第三届人类蛋白质组学国际会议上,Gorg 题为 2D PAGE:Challenges for Proteome Analysis 的讲座中提供了以下方法供参阅,见表 8.4 和表 8.5。

4. 等电聚焦
固相 pH 梯度等电聚焦的加样方法有两种。一种是在干胶条泡胀时将样品加在泡胀液中,样品浓度 5~10 mg/ml,样品体积 350 ul (180 mm 胶条 ),450 μl ( 240 mm 胶条)。另一种是在等电聚焦前加样品。样品可加在样品杯中,样品浓度 5~10 mg/ml,样品体积 20~100 ul,也可加在加样滤纸块上。用样品杯加样有利于碱性蛋白的分离,此时通常将样品加在阳极。具体操作请参阅仪器说明书。
固相 pH 梯度等电聚焦时电参数的设置通常采用恒功率的方式。由于 IPG 凝胶条的离子强度低,因而聚焦过程中电流很小(往往小于 1 mA )。当蛋白质分子向等电点方向迁移时,电流也逐渐减小,此时为了提高分辨率,必须加大电压。如采用恒功率电源,电压便会自动上升,通常高至数千伏。凝胶条的长度 pH 范围、样品的组成、泡胀液的组成,上样方式等都会影响电压的升高,其中样品中的盐对电压升高的影响最大。电压越高,聚焦时间越短。过长的聚焦时间会导致拖尾和损失蛋白,这种现象称为 过聚焦。
典型的等电聚焦程序有几个步骤,如在电泳槽上泡胀干胶条,所需电压很低 (几十伏)。加样时也采用低电压(200~500V),因为低电压有利于样品进入凝胶,并且能减少蛋白的聚合和相互作用。然后逐步分级升高电压,直到达到预置的电压(或伏·小时)并维持几个小时。见表 8.6,表 8.7 和表 8.8。
等电聚焦后的凝胶条可以密封在塑料袋或试管中,贮存于 -80℃ 数月。

5. pH 梯度的测定
由于 IPG 凝胶介质的导电性很低,不能用表面电极测定,如需要可用双向电泳蛋白标准在第二向电泳后测定。如果是宽 pH 范围,也可以采用等电点蛋白标准在等电聚焦后测定。
6. 平衡
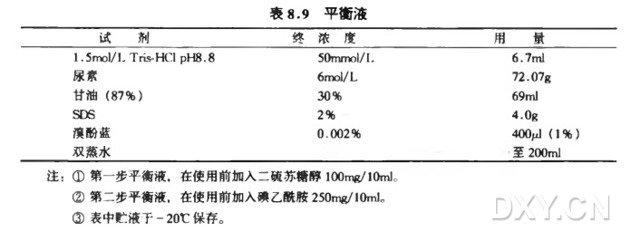
由于双向电泳的第一向固相 pH 梯度等电聚焦后的样品并没有满足第二向 SDS 电泳的条件,所以聚焦后的 IPG 凝胶条需要在平衡液中平衡。平衡分二步进行,先在含有 SDS、还原剂等但不含碘乙酰胺的平衡液中平衡(振摇 10~15 分钟),以将蛋白解聚成多肽链,并包裹负电,再在含碘乙酰胺的平衡液中平衡 ( 振摇 10~15 分钟),见表 8.9。碘乙酰胺的烷基化作用可保护 SH 基团,防止蛋白氧化,减少 纹理 (streaking ) 现象,得到清晰的图谱,参见 SDS 聚丙酰胺凝胶电泳。但平衡时间过长会丢失蛋白并使质谱分析复杂化。
平衡后用滤纸吸去多余的平衡液。将凝胶横向竖立在滤纸上,以免损失蛋白或损坏凝胶表面。
二、第二向
1. SDS 电泳
参照 SDS 聚丙酰胺凝胶电泳 配制合适的浓度梯度范围的 SDS 凝胶。
因为 IPG 凝胶条聚合在支持膜上,所以如果第二向仍然是水平电泳,只要将胶面朝下贴于 SDS 凝胶的浓缩胶的胶面上(阴极侧),避免气泡陷入。为了提高分辨率,当溴酚蓝前沿移出凝胶条 2 mm 时移去等电聚焦凝胶条,将阴极电极条移至凝胶条处后继续 SDS 电泳。如果第二向是垂直电泳,要用琼脂糖将凝胶条密封在浓缩胶上。先将琼脂糖在 100℃ 融化,再待之降到 60℃ 时慢慢地将凝胶条固定,同样要避免气泡陷入。为了以后分子质量的测定,在 SDS 凝胶的一端加分子质量标准。SDS 电泳方法请参阅 SDS 聚丙酰胺凝胶电泳。
2. 检测
考马斯亮蓝染色、银染色、荧光标记、放射自显影等方法均可用于双向电泳的检测,参见 常规聚丙烯酰胺凝胶电泳、SDS 聚丙酰胺凝胶电泳。
由于质谱检测对蛋白质质量的要求(5 ug ),所以虽然考马斯亮蓝染色的灵敏度较低,但仍然不失为双向电泳后常用的染色方法。
如果目标蛋白是低丰度蛋白,则应采用银染色,银染方法可参见 常规聚丙烯酰胺凝胶电泳、SDS 聚丙酰胺凝胶电泳,为了与质谱分析兼容,在敏化步骤中不加戊二醛,也不需要加甲醛,当然为此灵敏度有所降低,但也可检测到 5 ng 的蛋白。
锌-咪唑负染法检测极限为 15 ng,与质谱兼容性好,但定量不准确。
放射自显影和荧光成像是非常灵敏的检测方法,可低至 200 fg。为了进行放射自显影检测,可用同位素标记,一般使用 35S、14C、3H 和 32P。电泳后凝胶简单干燥,然后在 X 射线胶片或磷屏下曝光。荧光成像极其灵敏,凝胶电泳后与 PPO ( 2,4-diphenyloxazole ) 结合再检测。
使用荧光染料如 SYPRO、deep purple、Cydye 等进行荧光标记来检测不但灵敏度高,而且没有放射性,是当前最佳检测方法。特别是使用 Cydye 进行荧光差异分析简化了染色程序,并且提高了灵敏度、准确性和重复性,与质谱兼容性好,定量动力学范围宽。缺点是花费大,需要荧光扫描仪。
双向电泳后也可以转移到硝化纤维素膜或 PVDF 膜上以进行免疫化学检测或序列测定。
理论上,一块双向电泳凝胶能分离出多达 15000 个蛋白斑点,实际上,到目前为止最多可得到 11000 个蛋白斑点。大多数实验室到目前为止只能分离出几千个蛋白斑点。纵然如此,想用肉眼来比较双向电泳凝胶之间蛋白斑点的差异(新出现的斑点、斑点消失、位置变化、量的变化等)是不可能的。所以必须通过图像扫描,存储为图像文件进行分析。扫描时应选择合适的分辨率,大多 数情况下 300 dpi ( dot per inch) 比较合适。相对于凝胶而言,缩小的图像会导致图像质量的损失或无法分辨;而放大的图像则会由于多余的像素而出现内插值 ( interpolated values),甚至会出现 像素效应(pixellation),影响测定。因此扫描时图像与凝胶大小最好是 1:1,如果是考马斯亮蓝或银染色,采用灰度透射模式扫描。
图像分析软件用于评价和量化电泳结果。在蛋白质组学研究中,图像分析既要对上游各步的结果做出定性和定量的评价,将一个直观的蛋白质双向图谱数字化,还要为下一步的分析提供依据。特别是在比较研究中,需要用图像分析软件通过对正常和异常细胞、组织等的电泳图谱间的匹配,找出差异蛋白点,再结合图像分析数据与内部和外部数据库比较的结果做出结论。由此可见,双向电泳图像分析在蛋白质组学研究中是不可或缺的。
现在各大公司都有 2-DE 图像分析软件可供选择,如 Gellab-Ⅱ、Kepler、 BioImage、Melanie、PDQuest、ImageMaster 2D Elite 等,自动化程度更高的 Z3 和 Progenesis 也已推出。图像分析软件虽然界面、算法和功能各异, 但是过程大致相同,包括点检测、背景消减、匹配、分子质量与等电点校正、数据标准化等。
3. 蛋白斑点的后续分析
如果需要对图像分析后感兴趣的蛋白质进一步分析,首先要将这些蛋白斑点用人工或自动化仪器提取,然后进行酶解。现在有 4 种酶解方法将蛋白切成肽段:
(1) 凝胶内酶切(in gel Digestion);
(2) 电洗脱后在溶液中酶切(electroelution and digestion in solution);
(3) 电转移到膜上后在膜上酶切(electrotransfer onto a membrane and cleavage on the membrane);
(4) 在印迹过程中酶切(digestion during blotting)。
酶解后可以用基体辅助激光解析电离飞行时间质谱(Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry,MALDI-TOF-MS) 测定凝胶内酶切后多肽混合物的质量,获得肽质量指纹图谱(Peptide mass fingerprinting,PMF)。这一方法中用质谱分析的是蛋白质被酶切后的多肽混合物,而不是分析蛋白质本身,因为电泳后凝胶上的蛋白质很难从凝胶上洗脱下来进行质谱分析,而且仅用蛋白质的分子质量在数据库内检索鉴别是很不充分的。与之相反,多肽容易从凝胶上洗脱下来,而且一个蛋白质被酶切后生成的一小组多肽的质量,能提供足够的信息在数据库检索鉴定。近年来的研究进展显示,从凝胶上切下数百个蛋白质的斑点,进行凝胶内酶切,用 MADLI-TOF-MS 测定 PMF,到最后数据检索鉴定的整个过程,可以实现自动化,进行高通量的蛋白质组学研究。
蛋白质组学研究经典方法的步骤见图 8.6。
 展开 |
|---|

















