瑞德西韦中美新冠临床结果大相径庭 最终能否获批?
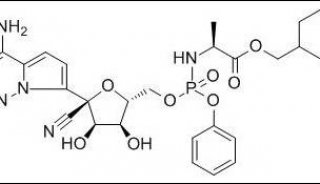
民间称瑞德西韦为“人民的希望”,中国从2月开始临床试验,美国的吉利德公司、NIH下属NIAID也分别开展临床实验。
4月11日,《新英格兰医学》杂志在线发布了瑞德西韦(Remdesivir)治疗新冠肺炎重症患者的首个临床研究结果,68%改善60%副作用,该药的中国重症临床试验已中止(原定4月27日)。4月29日晚,《柳叶刀》发表王辰、曹彬团队临床试验结果。研究显示,与安慰剂组相比,抗病毒药物瑞德西韦治疗危重症住院患者,并未加快新冠肺炎患者的恢复速度,也未降低病死率。中国的研究团队结果显示,瑞德西韦对治疗新冠肺炎无效。
而就在4月29日当日,吉利德科学公司发布其针对397名患者的开放标签临床III期试验结果显示,瑞德西韦对新冠肺炎早期患者治疗效果显著,超过一半患者在两周内出院。
4月30日早上,吉利德突然宣布停牌说有重大消息公布,结果是美国NIH下属的国家过敏和传染病研究所(NIAID)组织的双盲试验显示,瑞德西韦(Remdesivir)对治疗新冠病毒确实有效。
为何中美结果大相径庭 ?针对美国的结果,早先发布结果的曹彬如何回应?
且听下文分解:
王辰、曹彬团队双盲研究显示无效 未能减少死亡
试验结果表明:瑞德西韦没有显著缩短临床改善所需时间,其中,瑞德西韦组中位临床改善时间为21天,安慰剂组为23天;而在接受治疗时症状出现不足10天的患者亚群中,瑞德西韦组的临床改善时间快于安慰剂组,但未达到统计学显著性;28天临床表现改善率上,瑞德西韦组为65%,对照组为58%;瑞德西韦并未显著降低患者病死率,其中瑞德西韦组患者病死率为13.9%,对照组病死率为12.8%,且瑞德西韦未能有效降低患者的病毒载量。
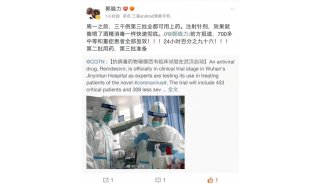
王辰、曹彬团队针对新冠肺炎重症患者的随机、双盲、安慰剂对照多中心试验在湖北10家医院开展,在今年2月6日至3月12日之间,共入组了237名患者,入组者均为经CT检查确诊的新冠病毒成人感染者(年龄≥18岁),并符合多项条件,包括从症状出现到入院的间隔均在12天以内,氧饱和度为94%甚至更低等。

患者按照2:1的比例随机分配至瑞德西韦注射组(第一天注射200mg,第2—10天每天注射100mg)和同等剂量的安慰剂组持续10天注射。在236位符合条件的入组者中,158例患者接受了瑞德西韦治疗,78例接受了安慰剂治疗。患者被允许同时使用洛匹那韦-利托那韦、干扰素和皮质类固醇,并分别在第1、3、5、7、10、14、21、28天收集粪便或肛门拭子样本,用以检测病毒RNA和定量。研究人员同时制定了一套标准,用以衡量患者的临床改善情况,具体包括出院(得分为1)到死亡(得分为6)等6种不同情况。根据该标准,如果患者的得分在经治疗后可减少2分及更多,或者出院,即视为临床改善。
根据该研究,在4月10日的最后一次随访后,研究人员发现,瑞德西韦组的临床改善时间与对照组并无明显差异,瑞德西韦组临床改善时间的中位数为21天,对照组临床改善时间的中位数为23天,两组在第28天的死亡率也很接近,瑞德西韦组共有22例死亡,占比为14%,安慰剂组有10例死亡,占患者的13%。
另据临床试验注册网站ClinicalTrials.gov,包括上述试验在内,瑞德西韦在中国进行的两项临床试验因无法招募到足够数量的患者已经终止。
吉利德SIMPLE临床试验:5天治疗方案可行 透露Positive data
4月29日,吉利德(GILD.US)官网终于公布了万众瞩目的瑞德西韦III期(open-label)临床试验数据,这一数据主要基于吉利德自己开展的主要针对重症COVID-19患者III期SIMPLE临床试验,吉利德并未明确表态该研究数据成功或是失败,只是表明,接受瑞德西韦5天疗程的患者与接受10天瑞德西韦疗程的患者的临床改善相似。
SIMPLE临床试验初步结果
SIMPLE研究显示,瑞德西韦10天给药治疗方案和5天给药治疗方案显示出相似的临床改善,两组重症患者的中位临床改善时间分别为10天与11天,除意大利外,两个治疗组中第14天的总死亡率为7%。
该试验评估了对重症COVID-19住院患者给与瑞德西韦5天或10天的安全性和有效性。研究结果表明,接受5天瑞德西韦治疗的患者与接受10天瑞德西韦治疗的患者在临床状况的改善上较为相似(Odds Ratio: 0.75 [95% CI 0.51 – 1.12] on Day 14)。在两个治疗组中均未发现新的安全性信号。
在本研究中,在5天治疗组中50%患者的临床改善时间为10天,而在10天治疗组中50%患者的临床改善时间为11天。两个治疗组中均有超过一半的患者在第14天出院((5-day: 60.0%, n=120/200 vs.10-day: 52.3% n=103/197; p=0.14)。在第14天,5天治疗组64.5%(n=129/200)的患者和10天治疗组中53.8%(n=106/197)的患者达到临床痊愈。
瑞德西韦临床疗效与安全性数据
同时,该研究表明瑞德西韦在5天和10天治疗组中通常耐受良好。两组中超过10%的患者发生的最常见不良事件(AE)为恶心(5-day: 10.0%, n=20/200 vs. 10-day: 8.6%, n=17/197)和急性呼吸衰竭(5-day: 6.0%, n=12/200 vs. 10-day: 10.7%, n= 21/197)。7.3%(n=28/385)的患者出现3级或3级以上肝酶(ALT)升高,其中3.0%(n=12/397)的患者因肝功能检查升高而停止瑞德西韦治疗。
针对以上研究,吉利德官方仅仅是客观描述了该研究取得的数据,并没有进一步分析其临床成功与否,美国NIAID(过敏和传染病研究所)公布的数据表明其开展的另一项瑞德西韦COVID-19临床试验(该研究为随机双盲安慰剂对照的临床试验)取得了“Positive Data”。这表明瑞德西韦针对COVID-19的临床试验很有可能获得成功。
吉利德科学计划在未来几周内提交完整的数据,并在同行评审的期刊上发表。
另据报道,美国国家过敏症和传染病研究所(NIAID)公布的数据显示,接受该药物10天治疗与5天治疗的患者临床状态均出现相似改善,第二次简单试验的前600名患者的结果预计在5月底出炉。吉利德科学首席医疗官Merdad Parsey博士表示,前述研究结果是对NIAID进行的针对瑞德西韦的安慰剂对照研究数据的补充,这有助于确定瑞德西韦的最佳治疗疗程。该研究表明,对部分患者来说,为期5天的治疗方案或许可行,这无疑将显著扩大在瑞德西韦目前可供应量内有望接受治疗的患者数量。
值得注意的是,吉利德官网第二条消息“Gilead Sciences Statement onPositive Data Emerging From National Institute of Allergy and InfectiousDiseases’ Study of Investigational Antiviral Remdesivir for COVID-19”透露出,由美国NIAID(国立过敏和传染病研究所)开展的另一项瑞德西韦COVID-19临床试验(该研究为随机双盲安慰剂对照的临床试验)取得了“Positive Data”。
吉利德披露信息
美国NIAID临床试验:31%的患者用药后症状改善
美国(NIH)下属美国国家过敏和传染病研究所(NIAID)对全球大约1090名患者展开的随机双盲对照临床试验是迄今为止最重要和最严格的,也是规模最大的试验,而且是美国政府机构的测试结果。试验采用双盲组(即同时采用药物和安慰剂),患者和医生都不知道谁得到了药物而不是安慰剂,这意味着无意识的偏见不会影响结果,而且这项试验的主要目的是看患者的病情需要多久能有所改善。
NIAID主导ACTT研究数据则显示达到了研究的主要终点,结果显示,31%的患者在用药后症状出现改善。接受瑞德西韦治疗患者的中位恢复时间为11天,安慰剂组患者的中位恢复时间为15天。治疗组的恢复时间显著缩短了31%;生存获益上,瑞德西韦组的死亡率为8.0%,而安慰剂组的死亡率为11.6%(p = 0.059),未达到统计学意义。
福奇在白宫记者会上简单介绍了这项临床试验结果,称其证明了瑞德西韦可阻断病毒。
美国NIH 还公布了有对照组的实验结果,和使用安慰剂的有严重肺部症状的重症住院病人相比,使用瑞德西韦的病人恢复时间加快,这项实验自2月21日开始,一共有1063个病人参加,一家独立的数据分析和安全监控委员会(DSMB)于4月27日开会审查并分析了测试的数据结果,认为接受瑞德西韦药物病人的恢复速度要快31%,使用该药物病人的中线恢复时间(能出院保持正常生活)是11天,而没使用药物的中线天数是15天,死亡率也有降低(11.6%降到8%)。
为何有不同的结果?曹彬如此回应:
针对中美临床试验的结论为何出现截然相反的结果,曹彬教授对记者表示:“这是两项不同的研究,评价标准不一样。”“我们对用药时机有要求,时间窗口在症状出现后的12天以内,但最主要的不同是研究终点的不同,美国NIH的研究终点指标过松。如果我们使用这一指标,估计也是阴性结果。”
据了解,王辰院士全程指导参与了这项试验设计与进行,试验设计非常完善、在研究过程中执行了最严格的标准、实验结果可信度也是最高的。
据第一财经日报报道,美国NIH和中国发表在《柳叶刀》的研究与设计均为双盲安慰剂对照临床(RCT),用药方案相同。“但是客观讲,中国瑞德西韦研究设计更加严格,科学性更强。”曹彬教授告诉第一财经记者。从主要终点来看,美国NIH设计的指标为临床恢复时间,中国则是设计了基于6分量表的临床改善时间。“NIH的恢复定义比较宽泛,包括住院,但不需要氧疗、出院(但可能仍有活动受限、需要吸氧),相当于我们的1-2级+回家吸氧。”曹彬教授表示。
对于这一设计,曹彬曾在公开场合多次表示,中国的两项瑞德西韦临床研究与美国NIH、WHO的试验设计相比,最大的差别就是在治疗窗口期方面用了非常严格的标准,而之所以这样设计,是因为他们发现在12天之内药物介入对于治疗新冠肺炎效果显著。
曹彬表示:武汉金银潭医院研究与ACTT研究终点不一样,是两个完全不同的研究,无法直接进行比较。而两个研究差异其实远不止在研究终点上。
武汉金银潭医院研究原计划入组453名患者,最终仅入组237名患者,他们被按照2:1比例分配至瑞德西韦组和安慰剂对照组。采取的研究主要终点指标为临床改善所需时间,定义为患者从入组接受治疗起28天内临床状态级别降低两级所需时间(患者临床状态评估为一个从死亡到出院的6分复合指标),但该研究由于未达计划入组人数,被提前终止。
ACTT研究的主要终点指标则为患者29天内的“临床恢复时间”,临床恢复包括三种情况:1,住院,不需要氧气支持;2,不住院,行动受限或需要氧气支持;3,不住院,行动不受限(不需要氧气支持)。同时设置8分量表作为重要的次要终点指标。
从患者入组标准上看,武汉金银潭医院研究要严格很多,要求均为重症(有影像学依据、血氧低于94%)患者且30天内未接受其他试验性药物治疗;ACTT研究仅要求满足有影像学依据、血氧低于94%、需要给氧、需要呼吸机四个条件之一即可入组,对前期治疗无要求。
而在治疗手段上实际上也存在差异,虽然双方对于瑞德西韦组都是进行10天给药,第1天静脉给药200mg初始剂量,接下来9天每天静脉给药100mg,但金银潭医院研究中还允许患者同时使用洛匹那韦/利托那韦、干扰素和糖皮质激素,这也成为研究的一个潜在影响因素。因此,也有外界声音对金银潭医院研究中对照组12.8%的病死率提出了疑问。
入组人数也会对研究结果及其效力产生影响,在一些小规模临床试验中由于样本数量小,一些数据差异可能无法得出统计学意义,但相同的研究可能会在样本量更大的研究中达到统计学意义。
如在ACTT研究中,瑞德西韦治疗组死亡率为8.0%,虽然相较于安慰剂组的死亡率11.6%有所降低,但是p值为0.059,未达到统计学意义。而考虑到这只是一项中期分析数据,目前后续研究仍在进行中,整个ACTT研究在死亡率这一重要指标上能否达到统计学意义,后续依然是可以期待的。
考虑到吉利德在4月中对数个瑞德西韦临床试验进行调整,相继扩大了入组人数,也有声音是猜测,吉利德在看到武汉研究的数据后,由于初步数据不及前期预计,进而在其他研究中扩大样本量以增强统计学效力。
实际上,除了样本量外,入组患者年龄、病情、基线水平等都会影响着不同数据的统计学效力,进而影响整个研究的效力。吉利德方面此前称,金银潭医院研究因患者太少而提前停止,因此无法得出有统计学意义的结论。
综合来看,和曹彬的研究相比,NIAID的研究在患者入组标准和终点设计上,都显得更加宽松,并且获得了较大的样本量支持,最终得出了阳性(positive)的研究结论。
瑞德西韦是否会获批?
虽然因为种种因素,两项研究不应拿来做“头对头”比较,但从数据上看,几项研究也提示了瑞德西韦在新冠肺炎治疗的一些共同趋势。如治疗组的临床改善时间均有所缩短,而对死亡率影响不显著。加上吉利德同日公布的开放标签III期SIMPLE研究数据,还可以看出瑞德西韦对于更早接受治疗的新冠肺炎患者有着更好疗效的趋势。
基于以上多项研究的数据,就目前美国国内以及除中国外全球严峻的公共卫生形势来看,FDA针对瑞德西韦COVID-19适应症的批准使用有两种可能。
1、直接走正式批准的流程,批准瑞德西韦以COVID-19适应症上市。但这一可能有两个问题,一是即使以最快的速度审评,按照繁琐的药物审批流程也需花不少时间才能最终完成审批;二是该研究毕竟不是双盲对照试验,对其数据的有效性还是存在疑问,在中国开展的双盲对照试验已经终止或暂停,而由NIAID开展的随机双盲安慰剂对照的多中心临床试验(NCT04280705)预计在五月下旬才能公布结果。因此,正式获批可能要等到NIAID的双盲临床试验公布后。
2、直接给与瑞德西韦紧急使用授权(Emergency Use Authorization,EUA),在同时继续推进瑞德西韦在美国及全球的临床试验。这一流程可以使广大患者不必再通过同情用药流程或扩大可及方案,也可以最快的速度用上瑞德西韦。EUA指美国FDA在实际的或潜在的紧急状态下对未获批准的医药产品及已获批准医药产品的未获批准用途的授权。在2009年H1N1流感期间,FDA就授予了当时尚未获批的静脉注射用帕拉米韦EUA用于临床防治严重流感;不久前,FDA又批准磷酸氯喹与硫酸羟氯喹作为EUA使用,用于治疗COVID-19患者。
相对来说,在公共卫生事件形势如此严峻的情况下,瑞德西韦作为EUA使用的可能性更高一点。关于这一点媒体也报道说,FDA计划最早在星期三宣布对瑞德西韦的紧急使用授权。
NIAID所长安东尼·福奇(Anthony Fauci)表示,瑞德西韦将被纳入美国新冠肺炎标准治疗中,而根据《纽约时报》报道,美国食药监局FDA已与吉利德就瑞德西韦开展沟通,将会尽快批准其用于新冠肺炎治疗的紧急授权(EUA)。
-
焦点事件

-
项目成果

-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件
-
焦点事件
-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件