







血红蛋白病及其检验
1)定义 血红蛋白病是一组遗传性或基因突变所致的血红蛋白合成障碍性疾病。根据其缺陷的不同又分为珠蛋白肽连数目合成异常或结构异常两大类。前者被称为珠蛋白生成障碍性贫血, 是常染色体隐性遗传性疾病,包括常见的β-珠蛋白生成障碍性贫血和少见的α-珠蛋白生成障碍性贫血;后者被成为狭义的血红蛋白病,为常染色体显性遗传性疾病,如HbS、HbE和HbC等。
血红蛋白病代表了一种世界范围内最常见的遗传性疾病。珠蛋白生成障碍性贫血约有1.5亿基因携带者;HbS约有1亿基因携带者;HbE在南亚约有30%的基因携带者;HbC在西非约有25%的基因携带者。
各种血红蛋白的组成见表8-7。
2)β-珠蛋白生成障碍性贫血 由于β链合成减少,过多的α链:①使含α链的血红蛋白HbA2或/和HbF的增加;②在幼红细胞和红细胞中形成包涵体,使红细胞特别扁平,中间的血红蛋白稍多,而周边较少,形成靶形红细胞;③形成的α链包含体附着于红细胞膜,使红细胞僵硬,导致无效造血和溶血;④使红细胞对钾离子的通透性增加,能量代谢能力降低,生存期缩短。
根据β-链合成是部分或是完全缺如,分别称为β+-和β0-两种基因型。其基因型和表型(或疾病)之间的关系见表8-8。

[诊断和鉴别诊断]
①重型β珠蛋白生成障碍性贫血:Ⅰ.多在出生后6~9个月出现贫血,发育障碍,智力受损,肝脾明显肿大,黄疸,骨骼改变,特殊“地中海贫血面容”。需要输血治疗;Ⅱ.血红蛋白<70g/L,MCV48~72fl,;靶形红细胞在10%~30%,网织红细胞5%~15%,外周血可见有核红细胞;骨髓红细胞系统明显增生;HbA为零或存在,HbF20%~95%,HbA2在纯合子时可以低、正常和增高。酸洗脱试验显示HbF分布相当异质

性;Ⅲ.双亲均为杂合子β+或β0珠蛋白生成障碍性贫血;或一方为杂合子β+,另一方为杂合子β0珠蛋白生成障碍性贫血;Ⅳ.患者珠蛋白肽链合成速率显示β/α降低(0~0.3),基因分析为纯合子β+或β0珠蛋白生成障碍性贫血,或双重杂合子β0/β+。
有慢性溶血性贫血的临床表现、HbF增加、能排除HbF增加的其他疾病,重型β珠蛋白生成障碍性贫血的诊断即成立。
②中间型β珠蛋白生成障碍性贫血:Ⅰ.多在2~5岁时出现贫血,症状和体征较重型轻,不需要输血;Ⅱ.血红蛋白70~100g/L,MCV明显降低,成熟红细胞形态与重型相似;网织红细胞3%~10%;偶见有核红细胞;HbA为零或存在,HbF20%~80%, HbA2量可减低、正常或增高;Ⅲ.双亲为β+或β0或其他类型珠蛋白生成障碍性贫血的杂合子;Ⅳ.珠蛋白肽链合成速率显示β/α比值降低,基因分析可知突变基因类因。
中间型珠蛋白生成障碍性贫血的特点是一些病例检查结果像重型,临床表现又不像。另一些病例检查结果像轻型,但临床表现又较重。他们包含着多种遗传学上不相同的疾病。具备重型β珠蛋白生成障碍性贫血的基本的诊断条件,但不需要输血,血红蛋白维持在70g/L以上,可诊断为本型珠蛋白生成障碍性贫血。
③轻型β珠蛋白生成障碍性贫血:Ⅰ.可有轻度贫血,无黄疸,偶见脾轻度肿大,无明显骨骼改变;Ⅱ.血红蛋白>100g/L;轻度小细胞,低色素,网织红细胞2%~5%,外周血无有核红细胞,HbA2>3.2%, HbF1%~6%;Ⅲ.双亲一方为杂合子β+或β0或其他类型珠蛋白生成障碍性贫血;Ⅳ.患者为杂合子β+或β0或其他类型珠蛋白生成障碍性贫血。
HbA2轻度增加,能排除HbA2增加的其他原因和缺铁性贫血,本并病的诊断即成立。
④极轻型β珠蛋白生成障碍性贫血:Ⅰ.偶见轻度贫血,无黄疸,无肝脾肿大,多为其他疾病做血液学检查时发现;Ⅱ.血红蛋白正常,偶见红细胞呈小细胞低色素,网织红细胞1%~2%;外周血无有核红细胞和靶形红细胞;HbA97%,HbA2<3.2%, HbF<1%;Ⅲ.双亲一方为杂合子β+或β0珠蛋白生成障碍性贫血;Ⅳ.患者珠蛋白肽链合成速率显示α/β为1.3左右;基因分析为杂合子β+珠蛋白生成障碍性贫血
本型诊断一般根据双亲的一方是正常的,另一方是HbA2升高的轻型β珠蛋白生成障碍性贫血而进行推断。珠蛋白肽链合成速率测定或基因分析可确诊。
3)α珠蛋白生成障碍性贫血
α珠蛋白生成障碍性贫血是由于α基因缺失所致的α链合成速度明显降低或几乎不能合成。在胎儿期过多γ链聚合形成γ4,即Hb Bart’s。Hb Bart’s与氧的亲和力高,在组织中放氧极少,常导致胎儿窒息死亡,或因胎儿水肿综合征在围产期死亡。出身后,由于γ链的合成逐步转化为β链,过多的β链聚合形成β4,即HbH。但因为HbH一般在30%以下,出生后能存活和成长。HbH是一种不稳定血红蛋白,形成小量的红细胞内包涵体,并易沉积在红细胞内;同时这种红细胞膜的通透性增高,红细胞膜易破碎,使红细胞的生存时间明显缩短,出现慢性溶血性贫血和骨髓造血代偿性增加。
α链合成部分缺如称为α+基因,完全缺如称为α0基因。α+基因表示为(-α),α0基因表示为(--)。α珠蛋白生成障碍性贫血表型与基因型之间的关系见表8-10。

[诊断和鉴别诊断]
①血红蛋白Bart’s胎儿水肿综合征:Ⅰ.妊娠30~40周时胎儿在宫内死亡或出生后半小时内死亡。胎儿苍白,全身高度水肿,体腔积液;巨大胎盘,孕妇可有妊娠高血压综合征;Ⅱ.脐血血红蛋白约49g/L,成熟红细胞大小明显不等、异形性、嗜多色性和小细胞低色素性,网织红细胞明显增加,有大量的有核红细胞;Hb Bart’s>70%, HbH5%~10%, Hb Portland 10%~15%;Ⅲ.胎儿双亲均为杂合子α0珠蛋白生成障碍性贫血;Ⅳ.胎儿缺失4个α珠蛋白基因,基因型为--/--(纯合子α0珠蛋白生成障碍性贫血)。
水肿胎儿,Hb Bart’s>70%,能排除Rh或ABO血型不合所致的胎儿水肿,诊断即成立。
②血红蛋白H病:Ⅰ.出生后一岁左右逐步出现轻度贫血,轻度黄疸,肝脾肿大,骨骼改变,轻度“地中海贫血”面容;Ⅱ.血红蛋白70~80g/L,MCV、MCH均降低;新生儿:Hb Bart’s20~40%,HbH 5%~10%,其余为HbF;大于6g个月或成人可见大量的HbH细胞(结晶紫染色有包涵体的细胞),HbH10%~20%;少量Hb Bart’s或/和HbCS;Ⅲ.患者双亲一方为杂合子α+珠蛋白生成障碍性贫血,另一方为杂合子α0珠蛋白生成障碍性贫血;Ⅳ.患者基因型为--/-α(混合杂合子α+α0)。
血红蛋白电泳出现HbH带,能排除继发性HbH病,诊断即成立。
③轻型α珠蛋白生成障碍性贫血:Ⅰ.常无贫血;Ⅱ.轻度低色素和小细胞增多;新生儿脐血Hb Bart’s 2%~10%,出生一年后消失;之后,HbH不存在,HbA2<2%;MCV70~80fl;罕见红细胞包涵体;Ⅲ. 患者双亲中一方为杂合子α0或双方均为杂合子α+珠蛋白生成障碍性贫血;Ⅳ. 患者基因型为杂合子α0(--/αα)或纯合子α+(-α/-α)珠蛋白生成障碍性贫血。
④无症状α珠蛋白生成障碍性贫血:Ⅰ.没有临床症状和体征;Ⅱ.新生儿脐血Hb Bart’s 0%~2%,出生一年后消失,之后,血红蛋白组成正常;Ⅲ. 患者双亲中一方为杂合子α+珠蛋白生成障碍性贫血;Ⅳ.患者基因型为杂合子α+(-α/αα)珠蛋白生成障碍性贫血。
虽然确诊依靠基因分析,但临床上此型常是通过对家族成员的研究而推断出来的。
4)镰形细胞贫血
是世界上最常见的狭义血红蛋白病,好发于非洲裔的人群。该病是由于β基因的第6个密码子中的腺嘌呤被胸腺嘧啶所替换,使β链上第6位上的谷氨酸被缬氨酸替换,生成的血红蛋白变异体称为HbS。HbS在脱氧的条件下形成纤维状多聚体。当多聚体量多达一定的程度时(HbS>50%),红细胞即发生镰形变。镰形变的红细胞易在血管内外被破坏而溶血。镰形变的红细胞也使血液的黏滞度增加,血流缓慢,引起血管堵塞。堵塞的血管加重组织缺氧和酸中毒,导致更多的红细胞镰形变。
镰形细胞贫血有纯合子和杂合子两种基因型,其临床和实验室特征见表8-12。HbS与其他血红蛋白病的双重杂合子,如Hb SC、Hb SE、Hb SD等统称为镰形变综合征。
[诊断] ①阳性家族史;②血红蛋白电泳或层析分析发现HbS;③血涂片上观察到镰形细胞改变有助于诊断。
5)血红蛋白C病
是由于β链上第6位上的谷氨酸被赖氨酸取代后的血红蛋白,致病机理类似于HbS。其临床与实验室特征见表8-13。
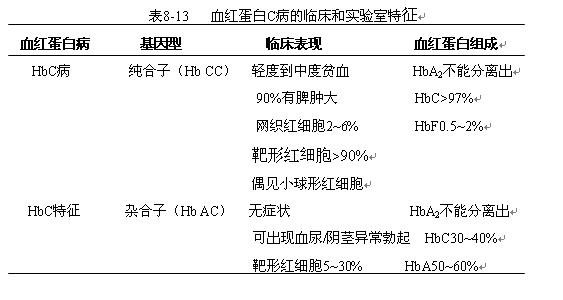 诊断依赖于血红蛋白电泳或层析发现HbC,阳性家族史很重要。
诊断依赖于血红蛋白电泳或层析发现HbC,阳性家族史很重要。
6)HbE病
是由于β链第26位上的谷氨酸被赖氨酸取代而形成的血红蛋白变异体,是第二种常见的血红蛋白病。80%的患者生活在东南亚。临床和实验室特征见表8-14。
诊断依赖于血红蛋白电泳或层析发现HbE。
7)不稳定血红蛋白病
是由于维持血红蛋白分子稳定性特别重要的氨基酸被替换所致,这主要是涉及到血红素与珠蛋白之间的连接处。其基因型都是杂合子,遗传方式是常染色体显性遗传。不稳定血红蛋白变异体可自发或被氧化性物质诱导而形成红细胞内的变性珠蛋白小体,使红细胞寿命缩短。这组疾病的临床表现为慢性或药物诱导的溶血性贫血,Heinz包涵体和中暗褐色尿。
热变性试验和异丙醇沉淀试验都能可靠地检测出不稳定的血红蛋白
>、Hb SD等统称为镰形变综合征。
[诊断] ①阳性家族史;②血红蛋白电泳或层析分析发现HbS;③血涂片上观察到镰形细胞改变有助于诊断。
5)血红蛋白C病
是由于β链上第6位上的谷氨酸被赖氨酸取代后的血红蛋白,致病机理类似于HbS。其临床与实验室特征见表8-13。
诊断依赖于血红蛋白电泳或层析发现HbC,阳性家族史很重要。
6)HbE病
是由于β链第26位上的谷氨酸被赖氨酸取代而形成的血红蛋白变异体,是第二种常见的血红蛋白病。80%的患者生活在东南亚。临床和实验室特征见表8-14。
诊断依赖于血红蛋白电泳或层析发现HbE。
7)不稳定血红蛋白病
是由于维持血红蛋白分子稳定性特别重要的氨基酸被替换所致,这主要是涉及到血红素与珠蛋白之间的连接处。其基因型都是杂合子,遗传方式是常染色体显性遗传。不稳定血红蛋白变异体可自发或被氧化性物质诱导而形成红细胞内的变性珠蛋白小体,使红细胞寿命缩短。这组疾病的临床表现为慢性或药物诱导的溶血性贫血,Heinz包涵体和中暗褐色尿。
热变性试验和异丙醇沉淀试验都能可靠地检测出不稳定的血红蛋白









