数字PCR技术进展简介
聚合酶链式反应 ( polymerase chain reaction,PCR) 提出至今已有20年时间,期间PCR已发展成为分子生物学领域的一项关键技术和常规技术,极大地推动了生命科学各个领域的发展。特别是 90 年代后期,美国 ABI 公司推出的实时荧光定量PCR( real time PCR,qPCR) 技术及相关产品更是将PCR由体外合成及定性/半定量检测技术发展成为一种高灵敏、高特异性和精确定量的基因分析技术。
尽管经过十几年时间的迅速发展,qPCR 技术已经用于除外伤和营养缺乏症外所有疾病的诊断,但是,在 PCR扩增过程中影响其扩增效率的因素有很多,不能保证在反应过程中扩增效率保持不变和实际样品与标准样品以及不同样品之间的扩增效率是相同的,由此导至其定量分析所依赖的基础——循环阈值(CT)不是恒定不变的。因此 qPCR 的定量只是“相对定量”,其准确度和重现性依然不能够满足分子生物学定量分析的要求。
20 世纪末,Vogelstein等提出数字PCR(digital PCR,dPCR) 的概念,通过将一个样本分成几十到几万份,分配到不同的反应单元,每个单元包含一个或多个拷贝的目标分子( DNA 模板) ,在每个反应单元中分别对目标分子进行PCR扩增,扩增结束后对各个反应单元的荧光信号进行统计学分析。与 qPCR 不同的是,数字PCR不依赖于CT值,因此不受扩增效率影响,扩增结束后通过直接计数或泊松分布公式来计算每个反应单元的平均浓度(含量),能够将误差控制在5%以内,数字 PCR 可以不需要对照标准样品和标准曲线来实现绝对定量分析。
该技术提出至今虽然只有十几年时间,但是由于其独特的技术优势和应用前景,使得其产业化发展相当迅速。迄今为止,已有包括Fluidigm和 Bio-Rad等几家公司相继推出了数字 PCR 产品,并已经应用于单细胞分析、癌症早期诊断和产前诊断等研究领域。目前,有关数字 PCR 技术的综述类文献并不多见,本文将在现有文献基础上,对该技术的原理、定量方法、分类及应用进行评述,并对发展趋势进行展望。
数字PCR技术的原理
1
数字PCR(也可称单分子PCR) 一般包括两部分内容,即PCR扩增和荧光信号分析。在PCR 扩增阶段,与传统技术不同,数字PCR一般需要将样品稀释到单分子水平,并平均分配到几十至几万个单元中进行反应。不同于qPCR 对每个循环进行实时荧光测定的方法,数字 PCR 技术是在扩增结束后对每个反应单元的荧光信号进行采集。最后通过直接计数或泊松分布公式计算得到样品的原始浓度或含量。
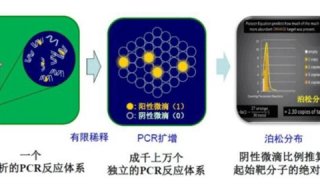
最初 Vogelstein等提出的数字 PCR 是在96孔板中进行的,如图 1 所示。DNA 模板被稀释成大约平均每两个孔内有一个拷贝的浓度,在经过优化的实验条件下进行 PCR 扩增。他们设计了两种带有不同荧光基团的分子信标探针分别与 PCR 产物杂交,其中一种探针可以与野生型和突变型两种产物杂交,另一种探针只与野生型杂交,通过直接计数每个孔内的荧光信号得到同一样品中等位基因(或野生型与突变型) 的数目和比值,并利用统计学方法分析样品间的显著性差异。目前的数字 PCR 技术主要采用分子信标和TaqMan探针两种方式对 PCR 产物进行荧光标记。其中分子信标法(如图1b所示) 通过一对通用引物得到包括野生型和突变型 在内的 PCR 产物,再经过不对称 PCR( asymmetric PCR) 得到单链 DNA 分子与两种荧光分子信标分别杂交,利用荧光颜色区别野生型和突变型,通过具有不同荧光反应单元数量的多少和比率进行分析,这种方法也被称为数字SNP( digital singlenucleotide polymorphism,digital SNP) 。TaqMan 探针法则可用于基因表达分析和单细胞多重 PCR等。Vogelstein 等提出一种基于磁珠和微乳液的固相数字 PCR 技术——BEAMing ( beads, emulsion,amplification,magnetics) ,原理如图 1c 所示。BEAMing 技术通过将引物化学键合在磁珠表面,再将单个磁珠与目标分子包裹在微乳液滴中进行 PCR 扩增,将野生型和突变型目标分子在磁珠表面进行复制。扩增结束后进行破乳,再利用流式细胞技术进行荧光计数。该技术还通过预扩增反应,提高了系统的灵敏度,适合用于低概率的等位基因突变分析。BEAMing 技术可通过固液分离除去多余荧光探针,降低背景干扰,可采用普通荧光探针代替高成本分子信标和 TaqMan 探针,降低成本。但是该方法需要将单个目标分子与单个磁珠包裹在同一液滴中,增加了操作的复杂性和难度,需要进行大量条件优化实验。此外, Zhong 等提出通过调节荧光探针浓度实现多重PCR的技术,实现了SMA基因拷贝数变异和 c.815A>G突变位点等五重PCR分析,突破了通常只有4个荧光通道的局限。
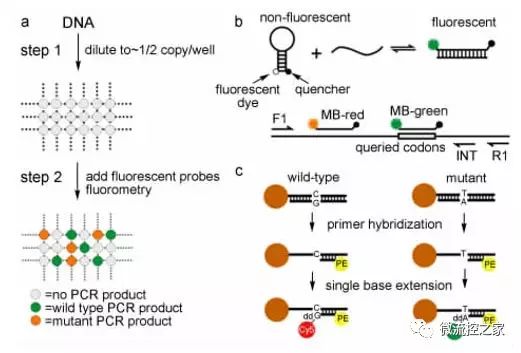
图 1 数字 PCR 技术原理: a) 数字 PCR 过程,第一步稀释样本分配至每个反应单元进行 PCR 反应,第二步荧光检测;b) 分子信标基因突变分析原理;c) BEAMing 检测原理
定量分析方法
2
传统的qPCR通常以循环阈值( cycle threshold,CT)为定量分析的基础,认为在指数扩增的开始阶段样品间的细小误差尚未放大且扩增效率恒定。对于一个 PCR 反应,到达循环阈值时
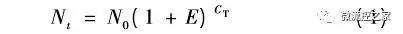
其中,N0 为初始模板的拷贝数,Nt 为第CT个循环时产物的拷贝数,E为扩增效率。将上式两边取对数,得到
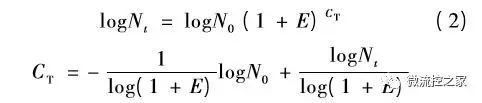
对于一个特定的PCR反应,扩增效率E和CT个循环时的拷贝数Nt均为定值,因此,CT值与初始模板拷贝数N0的对数成反比关系。然而,在PCR 扩增过程当中影响其扩增效率的因素有很多,比如酶和引物浓度等,因此很难保证扩增效率不变,导至定量PCR结果的准确度和精密度难以保证。
与传统qPCR方法不同的是数字PCR采用直接计数的方法进行定量分析,也就是在PCR扩增结束后有荧光信号(产物) 记为 1,无荧光信号(产物) 记为 0,有荧光信号的反应单元中至少包含一个拷贝的目标分子。理论上,在样品中的目标DNA浓度极低的情况下,有荧光信号的反应单元数目等于目标DNA分子的拷贝数。但是,在通常情况下,数字PCR的反应单元中可能包含两个或两个以上的目标分子,这时可以采用泊松概率分布公式( Poisson distribution) 进行计算。

上式中λ为每个反应单元中包含目标DNA分子的平均拷贝数(浓度),p为在一定的λ条件下,每个反应单元中包含 k 拷贝目标 DNA 分子的概率。λ由样品溶液的稀释系数 m 决定,有 λ= cm,其中c为样品的原始拷贝数(浓度) 。当 k = 0 (不含目标 DNA 分子) 时,上式可简化为 p = e^-λ = e^-cm, p 可以看作是无荧光信号的反应单元数与反应单元总数的比值,即

其中,n为反应单元总数,f为有荧光信号的反应单元数。上式两边取对数( ln) 得到
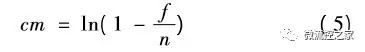
从数字 PCR 反应单元总数和有荧光信号的单元数以及样品的稀释系数,就可以得到样本的最初拷贝数(浓度) 。数字PCR的定量方法不依赖于扩增曲线的循环阈值,因此不受扩增效率的影响,也不必采用看家基因( house-keeping gene)和标准曲线,具有很好的准确度和重现性,可以实现绝对定量分析。
数字 PCR 的灵敏度也可以称之为分辨率,指的是对目标基因或突变的识别能力。它除了与检测器的灵敏度和PCR扩增效率等因素有关外,很大程度上取决于反应单元的数目n。理论上每个反应单元至多有一个拷贝的 DNA 分子,相同体积的情况下, n = 10^2 时,该方法的最大分辨率为 1 /100,也就是说样品浓度最低为1%可以被检出。如果 n = 10^4 时,就可以从10^4个分子中检测到1个靶标,即样品浓度最低为 0. 01% 可以被检出。因此,反应单元的数目越多,数字PCR的灵敏度越高,准确度也越高。
数字PCR技术分类
3
数字 PCR 技术提出至今,相关技术和产业化发展都非常迅速。迄今为止,数字 PCR 技术主要有三类: 微反应室/孔板、大规模集成微流控芯片和液滴数字PCR系统。
微反应室/孔板数字PCR
传统 PCR 或定量 PCR 反应都是在96/384孔板中进行的,因此早期的数字PCR技术也采用 96 /384孔板作为反应单元。但是数字PCR技术的灵敏度取决于反应单元的总数n,因此,理论上反应单元数越多越有利于提高灵敏度和准确度,普通的96 /384孔板无法满足检测的需要。而且,在96 /384 孔板中进行的PCR反应体系通常大于5μl,由试剂消耗引起的高成本问题令人望而却步。针对上述问题, Morrison 等在25mm×75mm不锈钢芯片上刻蚀了3072个直径为300μm的微反应室 (如图2a所示),每个反应单元的体积降低至33 nl。该芯片可在商品化 PCR 仪上使用,与384 孔板的检测灵敏度相当,但反应体积降低为原来的1/64,样品通量提高了24倍。随着反应单元数目的成倍增加,反应体积从微升级降至纳升级,传统的操作人员采用移液器加试样的方式已经无法满足快速精准取样的要求,因此需要借助高通量自动点样仪或机械手等设备,这无疑大幅提高了系统的成本和操作的复杂性。
大规模集成流路数字PCR
微流控芯片技术的发展为我们提供了一个实现低成本、小体积和高通量平行PCR分析的理想平台。2000年,Unger等采用多层软刻蚀 ( multilayer soft lithography,MSL) 技术在聚二甲基硅氧烷( polydimethylsiloxane,PDMS) 微流控芯片上设计并加工高密度微泵微阀结构(如图2b所示) ,他们将这种芯片称为IFC( integrated fluidic circuit)。IFC 利用PDMS材料具有高弹性的特点,通过多层软刻蚀技术在芯片上加工交织的液体和气体通道结构,可以快速并准确地将流体分成若干个独立的单元,进行多步平行反应。2006 年Ottesen等将IFC芯片用于数字PCR分析,通过精准控制微泵微阀的开启和关闭,一步操作即可将一个样本平均分配到 1176个反应单元中,每个反应单元的体积只有6. 25 nl,成功代替了传统点样仪和384孔板。他们同时进行了6个样本7 056个单元的平行数字PCR分析。此外, Hansen及其同事采用 MSL技术加工了具有10^6个结构单元的数字PCR 芯片,每个反应单元的体积降低至10 pl,芯片密度达到 440000 /cm2。与微反应室数字PCR系统相比,IFC的特点是通量更高,每个反应单元的体积更小,加样更快。最近,Men等在2mm×2mm区域内加工了82000个 fl 级反应单元,进行数字PCR分析。
液滴数字PCR
液滴数字PCR源于乳液PCR( emulsion PCR) 技术,即将DNA模板与连接引物的磁性微球以极低的浓度(比如单拷贝) 包裹于油水两相形成的纳升至皮升级液滴中进行 PCR 扩增,扩增后的产物富集在磁性微球上,收集破乳后进行测序。通过油水两相间隔得到的以液滴为单位的 PCR 反应体系,比微孔板和 IFC 系统更容易实现小体积和高通量,而且系统简单,成本低,因此成为理想的数字PCR技术平台。Vogelstein 及其同事提出的 BEAMing 技术就是一种基于乳液PCR的数字PCR系统。Lu 等也采用 BEAMing 和连接酶反应实现对 mRNA 的定量分析。Zhou 等在 BEAMing PCR 扩增后破乳,将连接不同产物的磁珠包被在聚丙烯酰胺凝胶中制成磁珠阵列进行荧光检测。但是,上述技术需要将单拷贝 DNA 模板与磁珠同时包裹在一个液滴中,增加了系统的复杂性和定量分析的难度。Beer 等在微流控芯片通道中用油相包裹皮升级液滴,液滴中只包裹了单拷贝 DNA 模板、 PCR引物及试剂,实现了数字PCR 定量分析。Hindson 等在微流控芯片中生成了20000—2000000个体积为 1nl 的液滴,然后将液滴转移到 96 孔板中进行TaqMan PCR扩增,至终点后将液滴从 96 孔板中取出,在微流控芯片中采用流式方法使液滴顺次经过双通道荧光检测器,以1000个液滴/s的速度进行计数。Pekin 等设计了一种微流控芯片,可分别生成包含不同荧光探针和 DNA 模板的液滴,再进行液滴融合,他们将该系统用于KRAS基因突变分析。此外,Shen 等提出了一种通过滑动芯片( SlipChip) 液滴数字PCR系统,设计了带有微流体通道和反应单元的玻璃芯片,上下两片之间用油相密封,通过滑动将样品溶液从流体通道中引入反应单元,同时生成1280个体积仅为2. 6 nl的液滴阵列,在芯片上进行PCR扩增和荧光成像分析。随后,他们还设计了具有不同体积反应单元的 SlipChip,从1 nl变化到125 nl,仅用不到 200个反应单元,理论上可以实现12000个等体积反应单元所能达到的检测浓度范围。Yang 等还合成了凝胶微球作为反应单元进行数字 PCR 扩增和定量分析,与前述液滴相比凝胶微球体系更加稳定和可控,易于收集和保存扩增产物。
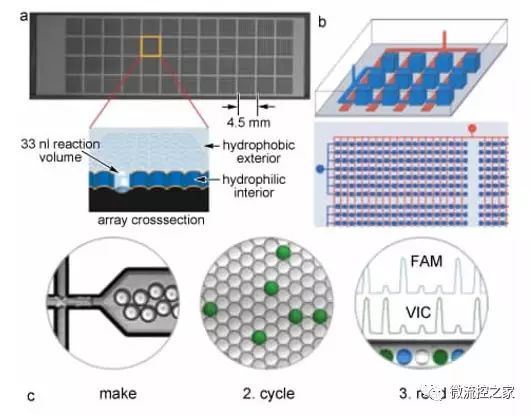
图 2 几种典型数字 PCR 芯片: a) 微反应室/孔板数字 PCR( OpenArrayTM); b) 大规模集成微流控数字 PCR 芯片( BioMarkTM); c) 微液滴数字 PCR( QX100TM)
数字 PCR 技术的应用
4
基因不稳定性分析
近年来随着人类对癌症的不断研究和认识,大量证据表明癌症是一种基因(染色体) 异常变化引起的疾病,普遍认可的异常情况包括癌基因及抑癌基因的突变、插入或缺失等。研究显示,癌症是由体细胞基因突变( somatic mutation),包括等位基因失衡( allelic imbalance,AI) ,杂合性缺失( loss of heterozygosity,LOH) ,拷贝数变异 ( copy number variations,CNVs) 等一系列基因变异情况引起的。 不过,癌细胞通常与大量正常细胞同时存在,因此,如何从大量正常细胞的 DNA 中检测到少量的异常基因成为癌症研究领域关注的焦点问题之一。Vogelstein及其同事提出数字 PCR 的初衷就是通过稀释将样品分配至尽可能多的单元中,从而将突变基因分子与大量的野生型基因分离开,在高背景条件下得到少量突变基因的信号。他们以KRAS基因突变为研究对象,对肠癌患者的粪便样品进行了数字 PCR 分析,得到 KRAS 基因第12号密码子点突变率约为4% (4/102)。接着,他们利用数字SNP技术研究了肠癌患者染色体18q杂合性缺失与血管侵入行为的关系,结果显示13个没有发生染色体 18q LOH的肿瘤样本均没有发现血管侵入行为,而 18个具有染色体18q LOH的肿瘤样本中有17个发生了癌细胞的血管侵入。Shih等研究了卵巢浆液性腺癌( ovarian serous carcinoma) 患者腹水中BRAF基因 599位密码子, KRAS 基因12和13位密码子基因突变情况,及其他 7 种标志物在包括卵巢癌、胰腺癌和肠癌患者体液中的基因突变情况。 Hanlon 等则对多发性骨髓瘤( multiple myeloma) 病人骨髓活检样本中染色体 13q 14缺失情况进行了研究,不经任何细胞富集技术,他们在 18 个病人中检测到16个LOH( 89% ) ,灵敏度高于传统荧光原位杂交( FISH) 技术。Yung 等考察了35个非小细胞肺癌临床血样中表皮生长因子受体( EGFR) 基因外显子 19 缺失及 L858R 突变情况,与直接测序技术相比,数字PCR技术在突变率低于30%的体液样本中具有更高的灵敏度和检出率。
人类基因组中存在一些范围从kb到Mb片段的亚微观结构变异,主要包括缺失、复制及嵌入等,统称为拷贝数变异( CNVs)。近年来的研究发现, CNVs与包括 HIV-1 感染在内的多种疾病密切相关,对于研究基因变异在生物进化、生理过程及疾病进程等领域具有重要意义。现有的CNVs研究技术包括比较基因组芯片(microarray based comparative genomic hybridization,array-CGH) ,高密度寡核苷酸芯片( high density oligonucleotide array,FISH) ,测序及定量PCR等,但是上述技术在灵敏度和分辨率方面存在缺陷,例如测序只适用于高于 30%的变异率检测,而定量PCR难以分辨低于2倍的CT差异,不足以满足CNVs分析的要求。Ramakrishnan及其同事建立了一种用于研究CNVs的数字PCR模式及统计学方法,通过直接计数目标基因与参照基因(拷贝数为1的基因,例如RNase P)的数目,计算比值,得到目标基因的拷贝数情况,最高分辨率达到15% 。他们研究了40位乳腺癌病人和8位正常人组织样本中HEBB2基因CNVs情况,其中14例癌症组织样本中HEBB2基因 CNVs 高于5。Wang 等对16个细胞系和20个肿瘤样本中EGFR CNVs、外显子19缺失及 L858R 突变情况进行了研究,他们在早期切除的肺癌肿瘤样本中检测到微量的对抗癌药物敏感的EGFR突变( 0. 02%—9. 26% ),而且没有发现EGFR基因的拷贝数变异情况。Hindson 等考察了人类基因组中MRGPRX1、染色体X和CYP2D6的CNVs情况,以及癌症病人HEBB2基因CNVs。