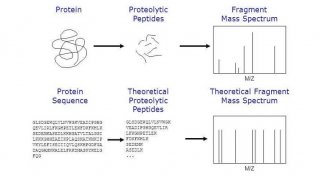
蛋白质测定方法及其比较
目前常用的有四种古老的经典方法,即定氮法,双缩尿法(Biuret法)、Folin-酚试剂法(Lowry法)和紫外吸收法。另外还有一种近十年才普遍 使用起来的新的测定法,即考马斯亮蓝法(Bradford法); 还有近来广为应用的蛋白定量方法——BCA法。其中Bradford法和Lowry法灵敏度较高,比紫外吸收法灵敏10~20倍,比Biuret法灵敏 100倍以上。定氮法虽然比较复杂,但较准确,往往以定氮法测定的蛋白质作为其他方法的标准蛋白质。6 O! d- J2 r) @; g* t& J4 m! I
这些方法并不能在任何条件下适用于任何形式的蛋白质,因为一种蛋白质溶液用这几种方法测定,有可能得出几种不同的结 果。每种测定法都不是完美无缺的,都有其优缺点。在选择方法时应考虑:①实验对测定所要求的灵敏度和精确度;②蛋白质的性质;③溶液中存在的干扰物质;④ 测定所要花费的时间。
一 凯氏定氮法
这种方法是1883年Kjeldahl发明,当时凯氏只使用H2SO4来分解试样,来定量谷物中的 pro,他只知用H2SO4分解试样,而不能阐明H2SO4分解有机氮化合物生成氨的反应历程,所以只使用H2SO4分解试样,需要较长时间,后来由 Gunning加入改进,他改进的办法是在消化时加入K2SO4使沸点上升,这样加快分解速度,因为温度由原来硫酸沸点的380上升到400℃,提高了不 到67℃所以速度也就加快了,凯氏定氮法至今仍在使用。
我们在检验食品中pro时,往往只限于测定总氮量,然后乘以pro核算等数,得到蛋白质含量,实际上包括核酸、生物碱、含氮类脂、叶啉和含氮色素等非蛋白质氮化合物,故称为粗pro。
1 凯氏常量定氮法:
不论常量、半微量以及微量定氮法它们的原理都是一样的,首先第一个步骤是消化:
(1)消化:样品与硫酸一起加热消化,硫酸使有机物脱水。并破坏有机物,使有机物中的C、H氧化为CO2和H2O蒸汽逸出,而pro则分解氮,则与硫酸结合成硫酸铵,留在酸性溶液中。
(2)在消化过程中添加硫酸钾可以提高温度加快有机物分解,它与硫酸反应生成硫酸氢钾,可提高反应温 度,一般纯硫酸加热沸点330℃,而添加硫酸钾后,温度可达400℃,加速了整个反应过程。此外,也可以加入硫酸钠,氢化钾盐类来提高沸点。其理由随着消 化过程硫酸的不断地被分解,水分的逸出而使硫酸钾的浓度增大,沸点增加。加速了有机的分解。但硫酸钾加入量不能太大,否则温度太高,生成的硫酸氢铵也会分 解,放出氨而造成损失。
为了加速反应过程,还加入硫酸铜,氧化汞或硒粉作为催化剂以及加入少量过氧化氢,次氯酸钾作为氧化剂。但为了防止污染通常使用硫酸铜。
所以有机物全部消化后,出现硫酸铜的兰绿色,它具有催化功能,还可以作为碱性反应指示剂。
(1)蒸馏:样液中的硫酸铵在碱性条件下释放出氨,在这操作中,一是加入氢氧化钠溶液要过量,二是要防止样液中氨气逸出。
(2)吸收与滴定:
蒸馏过程中放出的氨可用一定量的标准硫酸或标准盐酸溶液进行氨的吸收,然后再用标准氢氧化钠溶液反滴定过剩的硫酸或盐酸溶液,从而计算出总氮量。
半微量或微量定氮通常用硼酸溶液吸收后,再用标准盐酸直接滴定,硼酸呈微弱酸性,用酸滴定不影响指示剂变色反应,它有吸收氨的作用。
1.操作步骤:
准确称取样品中0.50-2.00g→于500ml凯氏瓶中→加10g无水K2SO4→加 0.5gCuSO4→加20ml H2SO4→在通风橱中先以小火加热,待泡沫消失后,加大火力,消化至透明无黑粒后,将瓶子摇动一下使瓶壁炭粒溶于硫酸中→继续消化30分钟→至到样液呈 绿色状态,停止消化,冷却→加200ml水→连接蒸馏装置→用硼酸作吸收液→在K氏瓶中加波动珠数粒和80ml50% NaOH→立即接好定氮球→加热→至到K氏瓶内残液减少到三分之一时,取出用水冲洗→用0.1N HCl滴定。
N(V2-V1)0.014
W
计算:
总氮量%= (N(V2-V1)×0.014)/W × 100
0.014----氮的毫克当量数
pro%=总氮%×K
乳制品K=6.38(N=15.7%)
小麦粉K=5.79(N=17.6%)
动物胶K=5.6(N=18.0%)
冰蛋K=6.7(N=14.8%)
大豆制品K=6.0(16.7%) K=6.25则(N=16%)
K-换称等数
各种试剂的作用:
浓H2SO4:
A :脱水使有机物炭化,然后有机物炭化生成碳,碳将H2SO4还原为SO2,本身则变为CO2
B: 氧化
C: pro与浓H2SO4生成NH3↑,CO2,SO2,H2O↑
D: NH3与H2SO4生成硫酸铵
(1)CuSO4的作用(催化剂)
CuSO4为红色沉淀,当C完全消化后,反应停止,红色消失,变为兰色,即为消化达到完全,兰色为CuSO4的颜色
(2)K2SO4的作用(提高沸点)
沸点由330℃提高到400℃加速了反应过程。
(3)硒粉和过氧化氢,氧化汞都为催化剂,但为了防止污染通常采用硫酸铜
(4)50%NaOH的作用
下面我就针对几点来说明为何影响氨化完全和速度快的因素:
(1)K氏烧瓶和取样量
如果称1g以上的样品,就需要K氏烧瓶最小500ml,800~1000ml的更好,这样的K氏烧瓶对于缩短氨化时间,加热的均匀性和完全氨化效果最好。
(2)分解剂
A H2SO4和K2SO4的添加量
有机无分解需要H2SO4量,H2SO4应根据有机物种类不同而加的量就不同,如果试样含脂类高,则加H2SO4多,为了提高分解温度,要大量添加K2SO4,但不能太多,也不能太少,太少则氨化不充分。K2SO4和H2SO4的添加比例是:
1g样品 K2SO4: H2SO4=7g:12ml
这种比例在国内外都使用,是公认的
还有一种比例: K2SO4:H2SO4=10:20ml
B 催化剂
用作催化剂的有Hg、HgO、Se,硒化合物,CuSO4、TiO2,对Hg,HgO有毒但结果 好,Se与CuSO4得到结果是一种,TiO2,的结果偏低,采用不同的催化剂则消化时间不同, HgO消化麦子为38,Se与CuSO4消化麦子55,TiO2消化麦子70,所以在给出测定结果时要注明催化剂的类型。
(3)热源的强度
消化时热源的强度同迅速消化和完全氨化关系很大,即便盐类K2SO4加得多,如果热源弱,也是没有意义的,热源过强导致H2SO4损失,使氨回收率低,另外K氏瓶的容量大小,颈部的粗细和长短等,也与热源的强度有关。
(4)氨的蒸馏和吸收及滴定
蒸馏有两种:
1.直接蒸馏(装置简便,准确性好)
2 .水蒸汽蒸馏
蒸馏加NaOH是50%,加的量为H2SO4量的4倍,硫酸量为12ml,则NaOH为12×4=48ml,而且一般高于这个理论值,即加到50~55ml,如果NaOH量加的不够就变成H2S, H2S是强酸,使颜色变红。
吸收液有:
1标准H2SO4 用标准碱返滴定,甲醛红指示剂
2 硼酸 用HCl进行滴定,混合指示剂
目前都用硼酸吸收液,用硼酸代替H2SO4,这样可省略了反滴定,H2SO4是强酸,要求较严,而硼酸是弱酸,在滴定时,不影响指示剂变色范围,另外硼酸为吸收液浓度在3%以上可将氨完全吸收,为保险期间一般用4%。
〈6〉实验注意事项
a.样品应时均匀的,若是固体样品应事先研细,液体样要混合均匀。
b.样品放入K氏烧瓶时,不要黏附瓶颈上,万一黏附可用少量水缓慢冲下,以免被检样消化不完全,使结果偏低。
c.消化时,如不容易呈透明溶液,可将K氏烧瓶放冷后,加入30%过氧化氢催化剂2~3ml,促使氧化。
d.在整个消化过程中,不要用强火,保持和缓的沸腾,使火力集中在K氏烧瓶底部,以免附在壁上的蛋白质在无硫酸存在的情况下,使氮有损失。
e.如硫酸缺少,过多的硫酸钾会引起氨的损失,这时会形成硫酸氢钾,而不与氨作用,因此当硫酸过多底物被消耗掉或样品中脂肪含量过高时,要添加硫酸量。
f.混合指示剂在碱性溶液中呈绿色,在中性溶液中呈灰色,在酸性溶液中呈红色,如果没有溴甲酚绿,可单独使用0.1%甲醛红乙醇溶液。
g.氨是否完全蒸馏出来,PH试纸检查馏出液是否为碱性。
h.向蒸馏瓶中加入浓碱时,往往出现褐色沉淀无。这时由于分解促进剂与加入的硫酸铜反应,生成氢氧化铜,经加热后又分解生成氧化铜的沉淀,有时Cu离子与氨作用生成深兰色的络合物。
i.消化剂绿色后继续消化30分钟即可。
2.K氏微量定氮仪法
3.K氏半微量定氮仪法 (2 、 3原理一样)
操作方法大同小异,半微量法消化后,定容100ml,然后吸25ml蒸馏吸收液吸收。
N(V2-V1)0.014
W*10/100
计算总氮%=(N(V2-V1)×0.014)/(W×10/100)×100 & nbsp;
对于微量定氮仪法,仪器有了改进,样液称样少,蒸馏消化液也少,其它基本一样。
4.K氏自动定氮法
原理与上面一样,仪器,采用K氏自动定氮仪:其装置内具有自动加碱蒸馏装置,自动吸收和滴定装置以及自动数字显示装置,消化装置:由优质玻璃制成的K氏消化瓶以及红外线装置的消化炉。
二 水扬酸比色法:
1.原理: 样品中的pro经H2SO4消化转化为铵盐溶液后,在一定的酸度和温度下与水扬酸钠和次氯酸钠作用生成有颜色的化合物,可以在波长660nm处比色测定,求出样品含氮量,计算蛋白质含量。
2.方法 (1)标准曲线的绘制
取6个25ml容量瓶编号 0 1 2 3 4 5 6
分别加空白酸 液 2ml
分别加磷酸盐缓冲液 5ml
稀释至总体体积至15ml
分别加水扬酸 钠 5ml
37C水 浴 15分钟
加入次氯酸 钠 2.5ml
37C水 浴 15分钟
取出试液于660nm下进行比色,绘标准曲线。
(2)样品处理:
准确称样0.20~1.00g→于K氏瓶中→加15mlH2SO4和5g催化剂→电炉上加热到沸腾后→加大
火力消化→直到出现暗绿色时→摇动瓶子→K氏瓶全部消化后→冷却→加水至250ml容量
瓶。
(3)样品测定:
准确吸取上述消化溶液10ml→于100ml容量瓶中→定容→准确吸2ml→于25ml容量瓶中→加
5ml磷酸缓冲液→以下操作与标准曲线绘制的步骤相同
并以试剂空白微对照,测得样液的光密度,从标准曲线查出其含氮量。
(4)计算
C×F
含氮%= ---------------------------× 100
W×1000×1000
C---从标准曲线中查出测定样液的含氮量(Ug)
F---样品溶液的稀释倍数
W---样品重量(g)
pro%=总氮%×K(K可为6.25,也可查)
3.注意事项:
A 当天消化液最好当天测定,结果重现性好,如果样液改至第二天比色就有变化。
B 当在PH和试剂适当范围内加入氯源后,颜色的显色和温度有关,应严格控制反应温度。
C 这种方法测定结果基本与K氏法一致。
4.试剂
(1)氯标液:称经110℃干燥2h的硫酸铵0.4719g→于烧瓶中定容10ml→此液1ml相当于
1.0mg氮标液→使用时配制成1ml相当于2.50Ug氮标液。
(2)空白酸液:称0.50g蔗糖→加15mlH2SO4和5g催化剂→与样品一样消化→定容250ml→
使用时吸收此液10ml→加水至100ml为工作液备用。
(3)磷酸盐缓冲液:称7.1g磷酸氢二钠→加38g磷酸三钠→加20g三九石酸钾钠→加400ml水
溶解→过滤→另称35gNaOH溶于100ml水中→冷至室温→缓缓地搅拌加入磷酸盐溶液中→加
入水稀释至1000ml备用。
(4)水扬酸钠溶液:称25g水杨酸钠和0.15g亚硝基铁氰化钠溶于200ml水中过滤,加水稀释
至500ml
(5)次氯酸钠溶液:吸4ml安替富民液,用水稀释至100ml
5 仪器
(1)分光光度计
(2)恒温水浴
三 紫外分光光度法
1.原理:
pro及其降解产物的芳香环基 ,在紫外区内对某一波长具有一定的光选择吸收,在280nm下,光吸收与pro浓度(3~8mg/ml)成直线关系,因此,通过测定pro溶液的吸光度,并参照事先用K氏定氮法分析的标准样品,从标准曲线查出蛋白质的含量。
2.试剂:
(1) 0.1mol/l柠檬酸水溶液。
(2)8mol/l脲[(NH2)2CO]的2NNaOH溶液。 脲的2N氢氧化钠液
(3) 95%乙醇
(4) 无水乙醚
3.仪器
(1) 751型的紫外分光光度计
(2) 离心机
4.操作方法
(1)标准曲线的绘制:准确称取样品.2.00g,置于50ml烧瓶中,加入0.1mol/l柠檬酸水 溶液30ml,搅拌30分钟使其充分溶解,用四层纱布过滤于玻璃离心管中,以每秒钟3000~5000转的速度离心10分钟,分别吸出上清夜 0.5,1.0,1.5,2.0,2.5,3.0ml于6个10ml容量瓶钟,每个容量瓶,8mol/l脲的氢氧化钠溶液充分摇2分钟(如果离心再次离 心),取透明液于比色皿中,在280nm下测定其吸光度。(做参比值)
以事先用K氏定氮法测定的样品中蛋白质的含量微横坐标上面的吸光度为纵坐标,绘制标准曲线。
(2)样品测定
准确称取试样1.00g→于50ml烧杯中→加0.1mol/l柠檬酸溶液30ml→搅拌10分钟→四层纱布过滤到离心管中→用8mol/L脲的NaOH溶液定容,摇匀后于280nm下测吸光度,从标准曲线上查出pro的含量。
计算: 蛋白质= C/W× 100
C----从标准曲线上查得的pro含量(mg)
W----测定样品溶液相当于样品量(mg)
说明:
a.此法运用于糕点,牛乳和可溶性pro样品,测定糕点时,应把表皮颜色去掉。
b.温度对pro水解有影响,操作温度应控制在20~30℃。
四 双缩脲法-皮尼克法
1 原理:
双缩脲在碱性条件中,能与CuSO4结合成红紫色的络合物。pro分子中含有肽链与双缩脲结构相似,也呈此反应。本法直接用于测定像小麦粉等固体试样的pro含量。但作为铜的稳定剂,酒石酸钾钠比甘油好些。小麦粉中的pro能直接地一边抽出一边进行定量。
2 试剂
⑴甘油作为稳定剂:取10ml10N KOH溶液,3.0ml甘油加到937ml水中,激烈搅拌,同时加入4%CuSO450ml。
⑵酒石酸钠作稳定剂,吸10ml10N KOH溶液和20ml25%酒石酸钾钠溶液加到930ml水中,激烈搅拌,同时加入4%CuSO4液40ml。
配制以上两种溶液、试剂,必须澄清,无氢氧化铜生成,否则重配。
3方法:样品测定
标准曲线绘制
准确称0.6g样品→使用试剂(1)
准确称0.5g样品→使用试剂(2)
假如用试剂(2)
(1)准确称样→0.5g→于80ml离心管→加1mlClC4→混合→加50ml酒石酸钾钠稳定剂→ 盖上盖子离心10min(4000转/分)→放置1小时→吸混合液15ml→于20ml离心管中→离心到完全透明→取上清夜5ml于→10ml容量瓶→加 水定容→于550nm处测定吸光度,从标准曲线上查出pro含量。
(2)标准曲线绘制
按样品测定方法,根据样品中pro含量,取离心澄清样液0.0 2.0 4.0 8.0 10.0ml于10ml容量瓶中→分别加水定容,按照样品测定其吸光度。
事先用K氏定氮法测定样品中pro含量为横坐标,以上述吸光度为纵坐标绘制标准曲线。