生成式AI加速抗病毒药物开发,提高药物发现潜力
新兴药物靶蛋白的抑制剂发现具有挑战性,特别是当靶结构或活性分子未知时。
IBM 研究院、牛津大学和 Diamond Light Source 公司的合作团队,通过实验验证了深度生成框架的广泛实用性,他们的框架在蛋白质序列、小分子及其相互相互作用上进行了大规模训练,不偏向任何特定目标。
研究人员在生成基础模型上进行了蛋白质序列条件采样,为两个不同的靶点设计小分子抑制剂:刺突蛋白受体结合域(RBD)和 SARS-CoV-2 的主要蛋白酶。
尽管在模型推断过程中仅使用靶序列信息,但在每个靶合成的四个候选物中,有两个在体外观察到微摩尔水平的抑制。最有效的刺突 RBD 抑制剂在活病毒中和试验中表现出针对多种变体的活性。这表明,即使在缺乏目标结构或结合物信息的情况下,用于加速抑制剂发现的单一的、可广泛部署的生成基础模型也是有效且高效的。
该研究以「Accelerating drug target inhibitor discovery with a deep generative foundation model」为题,于 2023 年 6 月 21 日发布在《Science Advances》。

从头分子设计具有挑战性
从头分子设计,即提出具有所需特性的先前未识别的化合物,是药物发现和材料工程应用中的一个具有挑战性的问题。
例如,寻找那些作为进一步设计候选药物的化学起点的抑制剂化合物,通常涉及采用对含有标准化合物或较小化学片段的文库进行高通量筛选,但这类方法的成功率在 0.5% 到 1% 之间,具体取决于筛选的文库大小(通常约为 10^4 个条目)和目标特征。
成功率低的部分原因是搜索空间巨大,目前估计涵盖 10^33 到 10^80 个可行分子,其中通常只有一小部分具有所寻求的特征;因此通过实验来依次筛选是不可行的。
除了需要数千次筛选实验之外,文库的初始选择通常还需要与已报道的配体结合的目标蛋白的详细结构信息,而这些信息通常不容易获得。最后,由于基础设施、化合物和试剂的成本,导致抑制剂发现可能非常昂贵。
因此,迫切需要一种更有效的方法,以便能够从广阔的化学空间(包括尚未合成的分子)中蒸馏出以前未识别的和有前途的分子。这种方法将能够对一小部分候选药物进行实验验证,从而以更少的时间和成本提高抑制剂的发现率。
DL可应对挑战但也有局限性
基于深度学习的生成模型有可能以「无规则」的方式发现具有所需功能的先前未识别的分子,因为他们的目标是首先学习已知化学物质的密集、连续表示(以下称为潜在向量),然后修改潜在向量以解码为看不见的分子。因此,此类模型提供了进入以前未探索的化学空间的机会,不受人类有意识偏见的限制。
然而,对于目标特异性药物样抑制剂设计的任务,必须使用「反向分子设计」方法,其中通过学习的化学表示的导航是由分子属性属性引导的,例如目标抑制活性和药物相似性。在针对先前未识别的靶标设计抑制剂的情况下,需要足够量的示范分子,而这可能是无法获得的,并且需要昂贵且耗时的筛选实验才能获得。
由于大多数现有的深度生成框架仍然依赖于从特定目标的结合剂化合物库中学习,因此它们限制了对已知和整体分子的固定库之外的探索,同时阻止了机器学习框架对先前未识别的目标的泛化。
因此,虽然一些使用深度生成模型进行目标特异性抑制剂设计的研究已经过实验验证,但尚未有报道称这些模型能够在不同的蛋白质靶点上发现经过验证的抑制剂,而无需获得详细的靶点特异性先前结合数据(例如靶点结合剂分子)。
一种新的深度生成模型
IBM 研究院、牛津大学和 Diamond Light Source 公司的联合团队,展示了基于深层生成基础模型的单一、统一的抑制剂设计框架在不同靶蛋白上的现实应用性。生成框架只需要更容易获得的目标序列信息来指导设计。此外,该工作考虑了(i)设计命中的脱靶结合,以考虑潜在的下游不利影响,(ii)即使在未知结合物的情况下也能识别命中,以及(iii)优先考虑易于合成的化合物。
「开发和验证这些方法需要时间,但现在我们有了工作流程,可以更快地生成结果。」 该研究的共同高级作者、IBM 研究院研究员 Payel Das 说,「当下一种病毒出现时,生成人工智能可能在寻找新疗法中发挥关键作用。」
论文的联合资深作者 Martin Walsh 表示:「生成与感兴趣的药物靶标具有高亲和力结合的初始化合物,可以加速基于结构的药物发现流程,并支持我们为未来的流行病做好更好准备的努力。」
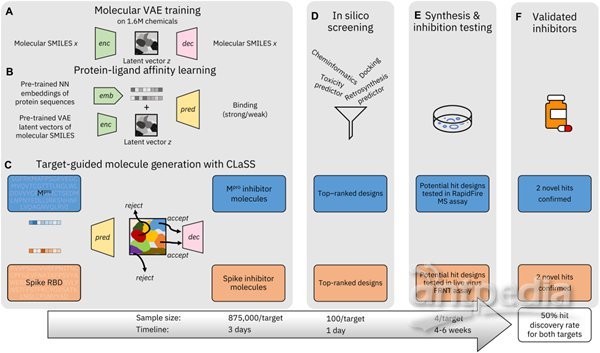
由 CogMol 驱动的抑制剂发现工作流程概述
研究人员使用 CogMol(一种深度生成模型)针对两个重要且独特的 SARS-CoV-2 靶标提出了先前未识别的且化学上可行的抑制剂设计:刺突 (S) 蛋白和主蛋白酶 (M^pro) 蛋白的受体结合域 (RBD)。
深层生成框架建立在化学分子、蛋白质序列和蛋白质-配体结合数据的大规模数据之上,作为目标感知抑制剂分子设计的生成基础模型,无需对特定目标数据进行任何进一步微调,并且可以外推到原始训练数据中不存在的目标序列。因此,CogMol 框架的这种广泛通用性将其置于新兴的「基础模型」类别中,这些模型是在大量未标记数据上进行预训练的,并且可以通过最少的微调用于不同的下游任务。
由 CogMol 设计的一组先前未识别的针对 SARS-CoV-2 蛋白的分子,于 2020 年 4 月在知识共享许可下在 IBM COVID-19 Molecule Explorer 平台上共享。在这里,研究人员通过合成和测试一些针对 S 蛋白和 M^pro 蛋白的 SARS-CoV-2 RBD 的优先设计的抑制活性,对 CogMol 深度生成框架的广泛实用性和就绪性进行了首次实验验证。
该团队对类先导化学物质库进行虚拟筛选,进一步证明了 CogMol 框架中使用的结合亲和力预测模型的适用性,并通过晶体学分析成功鉴定出三种化合物,证实其结合在M^pro的活性位点上,其中一种化合物表现出微摩尔抑制作用。
该研究首次提供了单一生成机器智能框架的经过验证的演示,该框架可以在设计过程中仅使用蛋白质序列信息,以高成功率为不同的蛋白质药物靶点提出先前未识别的有前途的抑制剂。
所设计的刺突抑制剂针对所关注的 SARS-CoV-2 变体表现出广谱抗病毒活性,进一步确立了这种深层生成框架加速和自动化命中发现周期的潜力,该过程已知产量低、损耗率高,但也增进了研究人员对较少探索的药物靶点的科学认识。
「我们使用生成基础模型为加速抗病毒药物的开发创造了有效的起点,而该模型对其蛋白质靶标知之甚少。」 该研究的共同高级作者、IBM 研究院研究员、牛津大学教授 Jason Crain 说,「我希望这些方法将使我们能够在未来更快、更便宜地制造抗病毒药物和其他急需的化合物。」