







机器学习加速探索材料的开发
设计空间几何增长是材料设计中的一大挑战。机器学习(ML)加速探索材料设计已经开始在的这一挑战中发挥作用,并显著提高了发现材料的效率。然而,这个流程暗含了密度泛函理论(DFT)产生的训练集的统计上的偏见。并且,在使用高通量计算产生训练集的时候,大量的计算会失败。这种情况对于一些有趣的,例如含有自由基,或者金属配位键的开壳层过渡金属功能材料和催化过程尤为显著。虽然这些材料非常吸引人,他们的复杂的电子结构和设计空间的几何增长使得该类材料设计异常困难。
在这篇Perspective中,Heather Kulik教授研究组描述了为改进传统基于DFT和ML的材料设计的流程所做的工作。通过搭建ML模型预测多个密度泛函给出的结果差别,促进了特定材料的敏感性分析。对于高通量计算所产生的的数据,发展了一系列的测试来确保每个数据点的高可靠性。最后,开发一系列机器学习的模型并且使用它们预测计算的成功与否和计算的体系是否含有强关联性,从而来决定是否需要对特定材料使用更加精确的电子结构方法进行计算。这些模型是迈向全自主化,不需要任何外在干预且稳定的,基于密度泛函理论的材料设计的第一步。
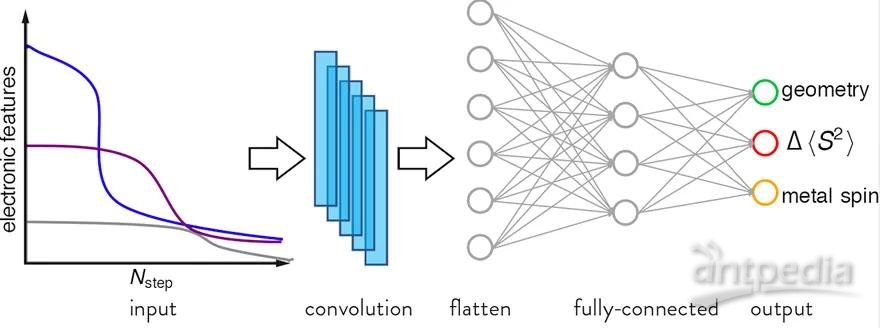
图1. 动态分类机的构架图。抽取计算的中间结果作为卷积神经网络的输入,从而预测多个影响最终计算成功与否的因素。
过渡金属配合物的计算常常面临失败的风险:从几何结构的角度考虑,最终优化的结构可能会有配位体脱离,第一配位层结构偏离的问题;从电子结构的角度考虑,因为DFT的最终波函数不保持S2,所以优化的波函数可能偏离我们想要的物理特性。一个计算中如果出现了这两种问题的任意一种,这个计算就是失败并且无效的。这些问题导致过渡金属配合物的几何优化计算的失败率很高,在特定体系下甚至达到50%,导致了大量计算资源和时间的浪费。
为了攻克这个问题,作者搭建了人工神经网络(ANN)模型,通过过渡金属配合物的化学构成和成键信息直接来预测这个结构的最终计算结果成功与否。研究发现这个静态模型可以在与训练练集相关的测试数据上表现的很好,有高于95%的准确率。这个静态模型只使用一半的计算耗费探索绝大部分(88%)的设计空间。但是这个静态模型的一个显著问题是:它的表现在面对与训练集统计上不相关的测试数据时会急剧下降。
考虑到在计算的过程中会产生很多与电子结构信息相关的中间变量,作者搭建了基于卷积经网络(CNN)的新模型。这个模型利用这些中间变量,动态的预测并且实时监测着每一个计算过程(图1)。随着计算的持续进行,这个动态分类机的准确率越来越高,远远超过之前的静态模型。由于使用了DFT的中间变量作为输入,这个动态模型的普适性强于静态模型,对于与训练集统计上不相关的测试数据也可以给出很好的表现。
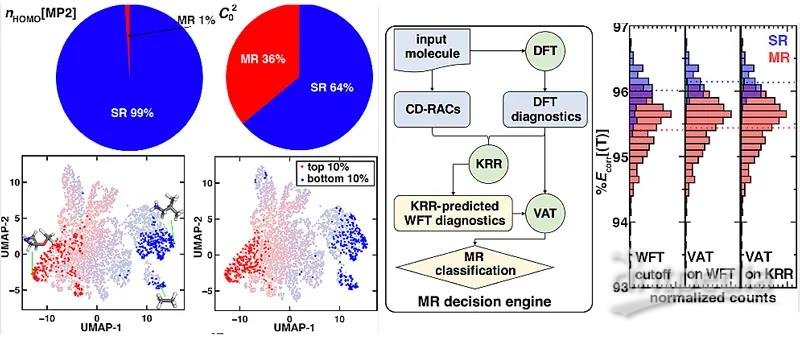
图2. (左) 根据nHOMO[MP2]和C02判断配合物是否具有强关联性。(中)强关联性分类机的流程图。(右)不同分类方法表现的对比。
过渡金属配合物的另一个显著问题是可能存在的强关联性。这是由于它们几近简并的d轨道能量决定的。如果一个过渡金属配合物拥有强关联性,这往往意味着DFT不能对它给出令人信服的描述。在理论化学的发展中,人们构造了许多基于不同电子结构方法的强关联效应诊断参数。但是,这些诊断参数之间的整体关联性不强,对于同一个体系,这些诊断参数很可能会给出不同的答案(图2,左上)。尽管如此,对于拥有极大或极小诊断参数的体系,所有的诊断参数却可以给出相似的答案(图2,左下)。因此,即使不能给所有的数据都令人信服的分类,我们依然可以很确信的对这些拥有诊断参数极值的体系加上标签(label)。
基于这个现象,作者搭建了一个半监督分类机(VAT):它既考虑到了这些有共同极值体系的标签,又考虑到了绝大部分无标签体系在整个诊断参数空间中的分布。在此过程中,发现基于波函数方法的诊断参数的计算量大却比较精确,于是作者搭建了一个基于DFT诊断参数和体系几何构性的核脊回归(KRR)模型来预测这些基于波函数方法的诊断参数。最终,将这两个模型结合在一起,构建了一个在DFT消耗下的强关联性分类机(图2,中)。研究发现,这个分类机相比较传统的无监督学习和化学上推荐的二分法,更加能够有效的区分有无强关联效应的体系(图2,右)。
最后,使用类似的机器学习模型,对大规模(十万个以上分子)高通量筛选中的电子强关联效应进行快速识别。这个模型能够在广阔的设计空间中迅速的找到“DFT安全岛”,将材料设计的重点放在这些有着较高DFT可信度的区域。
综上,作者总结了其研究组将机器学习分类机与高通量计算化学结合的几个工作。这些模型可以更加高效、中立、准确的产生基于DFT的数据,使得材料设计向全自主化,不需要任何外在干预且稳定的方向更近了一步。
-
焦点事件

-
焦点事件

-
会议会展

-
焦点事件





