
上海欧易生物医学科技有限公司

 分享至
分享至



分析解读 | 单细胞转录组测序技术揭示拟南芥根组织时空发育轨迹
前 言
单细胞转录组高通量测序是研究哺乳动物单细胞的常用方法,被广泛用于研究动物细胞组织分子异质性、细胞发育进程及稀有细胞类型鉴定等。随着液滴技术的发展和自动化程度的提高,单细胞 RNA 测序(scRNA-seq)的通量逐渐增高,并且在动物中被普遍使用,但在将其应用于植物时,诸如打破细胞壁的必要性(随后的转录效应)、高渗压敏感性和高细胞尺寸变异性等技术难题,仍然是植物单细胞面临的潜在挑战。
基本信息
文章名:Spatiotemporal Developmental Trajectories in the Arabidopsis Root Revealed Using High-Throughput Single-Cell RNA Sequencing
材料:拟南芥根组织原生质体
主要技术:Single-Cell RNA Sequencing
期刊:Developmental Cell
发表年份:2019.03.25
影响因子: 9.616
文章摘要
本研究利用单细胞转录组测序技术,对拟南芥的根组织进行了细致的研究和分析。作者首先利用常规 bulk RNA-seq 方法获得基因表达,与 scRNA-seq 技术的结果进行相关性分析,绘制了高分辨率的拟南芥根组织基因表达谱,并使用 seurat R 包对其进行 PCA 降维分析,利用 t-SNE 算法展示聚类结果。通过已发表的拟南芥根组织细胞类型的 marker 基因数据集和自创的无偏分析方法,揭示了根部主要细胞类型的特征。文章展示了如何区分 QC 细胞和分生组织细胞,并通过 monocle2 R 包进行拟时序分析,揭示了细胞从干细胞向分生细胞分化过程中所经历的复杂发展轨迹。与此过程有类似表达变化的高度互联的转录因子也表征了这一过程。总之,本研究提供了从时空视角研究拟南芥根组织细胞类型分化的方案。
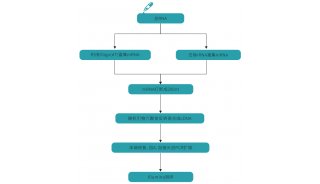
方法流程

研究内容
作者使用 scRNA-seq 技术和常规 bulk RNA-seq 对同样的样本进行了测序分析。通过两种方法的相关性分析,发现两种方法的基因表达具有很高的相关性(r=0.9)。另外, scRNA-seq 的两个生物学重复之间的基因表达相关性也很高(r=0.99),说明了 scRNA-seq 技术的准确性、可重复性和灵敏性,可以运用于植物数据分析。
通过 scRNA-seq 技术获得 4727 个高质量细胞的转录组数据,并使用 seurat R 包对其进行 PCA 降维分析,利用 t-SNE 算法展示聚类结果。为降低原生质体制备过程对转录组数据造成的影响,作者对原生质体(两个生物学重复)和非原生质体(两个生物学重复)分别进行了常规 bulk RNA-seq ,并使用 DESeq2 进行差异分析。在进行 scRNA-seq 数据分析之前,先去除由原生质体诱导的基因,最终鉴定出 15 个不同的簇。利用拟南芥根特异性 marker 基因集,作者确定了 8 个簇的细胞类群,例如簇 9 和簇 13 分别包含皮质和内胚层细胞。

图1. scRNA-seq 数据进行细胞聚类展示
作者分析了来自内皮层细胞报告基因系 pMIR166A:erGFP 材料的数据,发现被归类为内皮层组织的聚簇中的细胞确实有 GFP 的表达,证明此方法可信。对于 marker 基因表达不明确的细胞簇,作者进一步进行了分析。例如作者利用芯片数据集的 top10 差异基因确定 cluster 0, 1, 14 中多为混合的成熟细胞,而 cluster 2, 6, 7, 8 细胞的表达类似于分生细胞。

图2. 细胞特性和发育阶段在(亚)聚类中的反映
鉴于详细的参考数据集仅适用于极少数植物物种的特定组织和器官类型,作者开发了一种无偏方法来鉴定 scRNA-seq 数据产生的细胞簇的细胞类型。利用 seurat 包的 Bimod 算法对细胞簇进行差异分析,并定义PCT1(本细胞簇中表达该基因的细胞数)>10% 且PCT2(其他簇中表达该基因的细胞数)<10% 的基因为该簇的 marker 基因。通过对 marker 基因的 GO 富集分析,发现了与各簇细胞基因表达相关的生物学过程。为验证此无偏方法是否可信,作者选择了不同簇中的 10 个高特异性 marker 基因,使用转录启动子 3xVenus-NLS 报告序列进行验证,其中 8 个基因的表达与预测一致。

图3. 通过融合报告序列验证无偏的标记基因选择方法
为研究QC 细胞这一稀有细胞类型,作者对簇 11 进行了亚型分析,鉴定得到 36 个QC 细胞。通过 QC 细胞与分生组织细胞的差异分析,确认了 254 个在 QC 细胞中特异性表达的基因,其中一些未被报道过的 marker 基因(log FC >= 0.25; PCT1 >= 10%; PCT2 <= 10%)可能揭示 QC 细胞其他层面上的生物学功能。

图4. scRNA-seq 数据鉴定 QC 细胞 marker 基因的表达
为了揭示细胞从干细胞向成熟生毛细胞转变的过程,作者使用 monocle2 R 包对簇7, 5, 10的细胞进行了拟时序分析,利用 monocle2 R 包的差异 GeneTest 函数鉴定拟时序分析中有显著变化的基因。作者鉴定得到 3657 个高度分散的差异基因,使用 plot_pseudotime_heatmap 函数对这些鉴定到的基因进行了聚类,并使用 AtTFDB 数据库对转录因子进行注释。
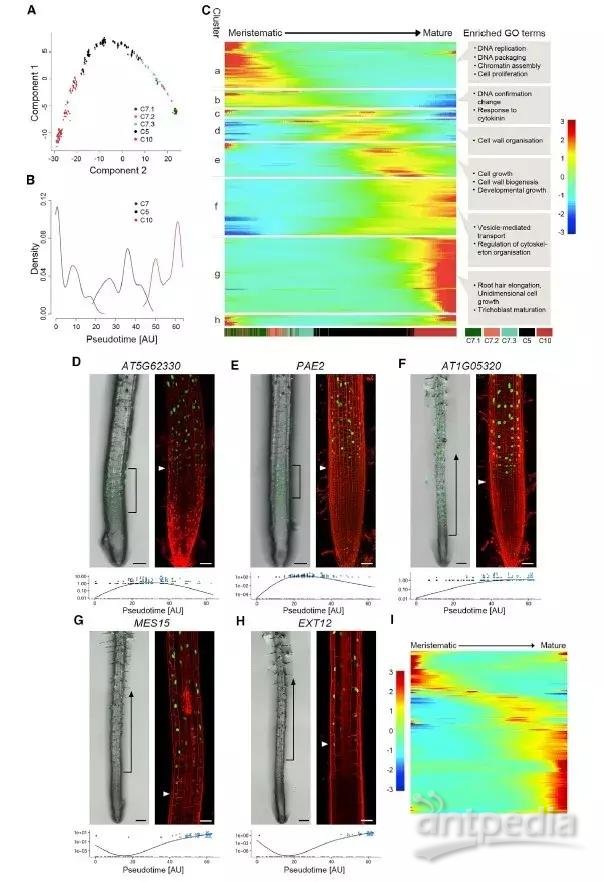
图5. 生毛细胞的拟时序分析
为进一步阐明生毛细胞分化过程中的遗传协调性,作者构建了拟时序分析中 239 个转录因子的基因调控网络。作者首先使用 monocle2 R 包的 genesmothcurve 函数对簇 h 中没有的转录因子的基因表达进行标准化,然后利用 SCODE 算法在 TFs 上推断基因动态调控网络,最后使用软件 Cytoscape 对结果进行可视化。从网络中可以清楚地看到,主要的、高度连接的中央调节器沿着整个轨迹存在,其中大部分没有涉及到根组织。将网络过滤到25 个核心组件,我们可以看到沿生毛细胞分化轨迹每一步的反馈调节的通道。

图6. 基因调控网络(GRN)预测生毛细胞分化过程中的关键调控因子
本文所描述的拟南芥根图谱提供了一个独特的根细胞类型分化的时空视角,对根的发育区域有了进一步的认识,并指出了大量参与这一过程的候选发育调节器。 scRNA-seq 技术将迅速成为植物科学中的一项核心技术,揭示越来越多的生物学秘密。
欧易单细胞测序平台介绍及分析结果展示
欧易生物是国内较早引入 10X Genomics 单细胞测序平台的机构之一,在农林及医学领域提供专业的单细胞建库测序及分析服务。凭借专业可靠的技术储备,已完成三十余种样本类型的成功上机检测及分析。目前,单细胞转录组分析内容包括降维、聚类,寻找 marker 基因,细胞类型鉴定,差异及富集分析,拟时序分析及个性化分析等。部分结果展示如下:

图7. tSNE降维聚类分析结果散点图

图8. marker 基因表达热图

图9. 不同类型细胞中 marker 基因差异表达小提琴图

图10. 细胞类型鉴定结果

图11. 基因表达量拟时序分析

图12. 细胞周期鉴定热图

图13. GSEA富集分析
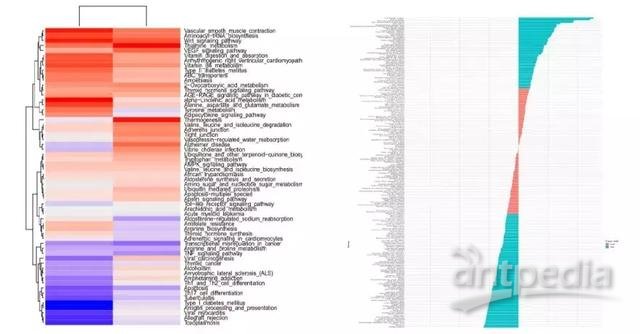
图14.GSVA富集分析
以上仅是部分展示,更多的高级分析比如加权基因共表达网络分析、单细胞调控网络等,等着各位老师进一步了解哦,快快联系欧易生物吧!
END
本文系欧易生物原创
转载请注明本文转自欧易生物
-
厂商文章

-
厂商文章

-
厂商文章
-
厂商文章

-
厂商文章

-
厂商文章
-
厂商文章

-
厂商文章
-
厂商文章

-
厂商文章

-
厂商文章

