上海欧易生物医学科技有限公司

 分享至
分享至



IF:29.886!单细胞转录组测序描绘肺癌T细胞免疫图谱
引 言
肠道微生态系统由肠道正常菌群及其所生活的环境共同构成。作为人体最庞大、最复杂的微生态系统,肠道微生物及其代谢产物不仅能调节人体健康,还可以在膳食营养和宿主先用几篇高分文献感受一下基于单细胞测序技术研究肿瘤免疫微环境这个方向的火热程度。

我们以肺癌为例,为大家解读单细胞测序在肿瘤免疫微环境中的文章内容,希望能够为研究肿瘤浸润淋巴细胞的老师提供思路。
基本信息

材料:来自于14个非小细胞癌患者的癌、癌旁和外周血的T细胞(共12,346个T细胞)
期刊:Nature Medicine
方法:Smart-Seq2
影响因子:29.886
研究目的:描述肿瘤浸润淋巴细胞的组成、谱系和功能状态的基本情况
研究背景及目的
肿瘤免疫治疗在非小细胞肺癌(NSCLC)中的效果不一,肿瘤浸润淋巴T细胞的组成部分决定了肿瘤免疫治疗的效果异质性。本文章的研究目的是通过单细胞测序揭示非小细胞肺癌中肿瘤浸润淋巴T细胞的组成、谱系以及功能状态,为临床非小细胞肺癌患者的分层分级提供新方法。
研究路线

技术框线示意图
研究内容
1. 非小细胞肺癌肿瘤相关T细胞的降维聚类分析
作者通过t-SNE降维和无监督聚类分析,将来源于癌、癌旁和外周血中的9,055个T细胞分为7个CD8+亚群和9个CD4+亚群(Fig. 1b),按照相关的marker,将这些cluster进行注释,如naïve、effector、resident memory以及exhausted等等(Fig. 1c)。此外,这些cluster有显著的组织分布偏好性。例如,CD8-C1-LEF1和CD4-C1-CCR7这两个naïve T cells亚群主要来源于血液中,CD8-C6-LAYN(exhausted T cells)和CD4-C9-CTLA4(Tregs)主要富集在肿瘤样本中(Fig. 1d)。
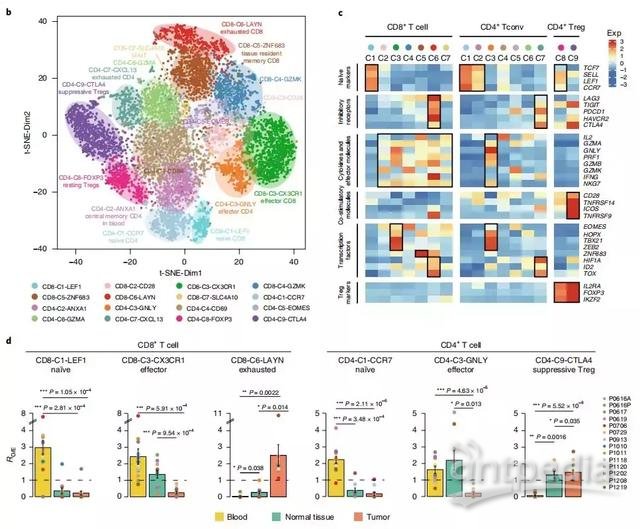
Fig. 1.非小细胞肺癌肿瘤相关T细胞的降维聚类分析
2. 单细胞免疫组库TCR克隆扩张分析

Fig. 2.通过TCRs确定的T细胞克隆扩张特征
下一步,作者试图通过免疫组库TCR信息,对T细胞进行分型。作者发现,效应T cell亚群CD8-C3-CX3CR1和CD4-C3-GNLY不仅克隆型的细胞占比最高(Fig. 2c),同时也有着较高比例的组织间克隆细胞(Fig. 2d),高水平的组织间分布意味着这些具有相同祖先的T细胞在血液和实体组织之间迁移。另一方面,作者发现这两个亚群高表达细胞黏附以及细胞迁移相关的基因,验证了这些T细胞的潜在迁移特性(Fig. 2e)。
3. 单细胞表达谱聚类结果和单细胞免疫组库分型结果联合分析
具有相同TCRs的细胞可以阐明不同T细胞cluster之间的关系。作者发现9%的CD8+ T cells克隆至少在两个亚群中出现(Fig. 3a),暗示这些CD8+亚群并不是独立存在,可能存在着状态转换。于是作者对CD8+ T cells做了一个拟时序分析,构建了亚群间潜在的发展轨迹(Fig. 3b,Fig. 3e),这一结果也通过亚群间TCR克隆分析得到验证(Fig. 3c)。随后作者重点关注CD8-C5-ZNF683这个亚群,拟时序的结果证明该亚群处在“预耗竭”(pre-exhaustion)阶段,非小细胞肺癌中该亚群的占比高于原发性肝癌(Fig. 3e)。Kaplan–Meier生存曲线表明该亚群的高表达和肺腺癌病人良好的预后有密切关联(Fig. 3f)。

Fig. 3.CD8+ T cells状态转换轨迹变化
4. 肿瘤中Treg的活化和免疫治疗靶点分析

Fig. 4.肿瘤中Treg的活化和肿瘤治疗靶点基因表达
接下来,作者聚焦到CD4+的CD4-C9-CTLA4 Treg亚群。作者发现抗原特异性Treg标志物TNFRSF9在CD4-C9-CTLA4中呈双峰分布(Fig. 4a),可能暗示着肿瘤中Treg激活和静止状态的两种分布。相对于TNFRSF9 - ,TNFRSF9 + Tregs中高表达260个基因(Fig. 4b),TCGA生存曲线结果说明着260个基因预示着较差的预后(Fig. 4c)。另一方面,作者通过TCGA肺腺癌中肿瘤浸润淋巴细胞的表达特征,将肿瘤浸润淋巴细胞分为两组(Fig. 4d)。第一组主要富集“预耗竭”CD8+T细胞、静止的Treg和激活的CD4+细胞,第二组主要富集耗竭性的T细胞和激活性的Treg。特别地,第一组的预后好于第二组,说明T细胞的组成可以作为临床预后的标志物。最后,作者探究了肿瘤免疫治疗靶点基因在各亚群中的表达情况(Fig. 4e和Fig. 4f),发现目标基因在亚群表达特征各有不同,这些表达特征为免疫治疗以及病人分层分级提供新思路。
参考文献
PMID: 29942094 DOI: 10.1038/s41591-018-0045-3
- END -
本文系欧易生物原创
转载请注明本文转自欧易生物之间起到重要的桥梁作用[1],多数情况下肠道内的微生物与宿主之间是共生的关系。越来越多的研究表明,肠道微生物对宿主的免疫系统,及其它许多生理功能起到至关重要的作用[2]。肠道菌群结构的紊乱会诱发肠道疾病[3]。甚至一些看似与肠道系统关联不大的疾病(如肥胖、糖尿病、克罗恩病等)也与肠道微生物有着密切的联系[4,5]。这些紧密的联系,往往需要我们通过不同的分析手段去深入挖掘。
(一)
与环境因子等相关的肠道微生态研究
通常我们通过研究肠道微生物的群落结构来揭示其与疾病、免疫等之间的关系。除此之外研究肠道微生物与环境因子(这里环境因子包含pH值、温度等环境因子数据,也包括测定的代谢物含量、细胞因子数据等)之间的关联,可以从另外一个角度揭示微生物和环境因子之间的相关性。
根据微生物丰度的变化,以及环境因子数据,进行两者间的相关性分析。并以相关性热图的方式进行展示:

图片说明:上图展示微生物及代谢物间的相关性。蓝色表示正相关,红色表示负相关,颜色的深浅代表相关性的高低(相关性越高,颜色越深);圆圈的大小也表示相关性系数高低,相关性系数越高圆圈越大。如果相关性P Value大于0.05,则无圆圈展示。
此外可以根据显著性相关的微生物-环境因子进行网络关联分析,结果展示如下:

图片说明:每个圆圈代表一种微生物或环境因子,圆圈之间的连线红色表示正相关,绿色表示负相关。网络图可以直观地展示微生物和环境因子之间的互作关系。上图中相同颜色代表同一大类微生物(如上图为同一个Phylum门)或者同一大类代谢物。
(二)
趋势变化的肠道微生态研究
对于不同处理,如健康组(A)→疾病组(B)→给药组(C) 肠道微生物研究,筛选出多个比较组之间差异OTU或物种的交集(A-vs-B和B-vs-C的差异交集),随后观察这些筛选出来的OTU或物种丰度趋势,可以很好地反映出OTU或物种丰度随着不同处理而变化:

图片说明:热图中颜色表示各个样本OTU丰度的高低(橙色表示相对丰度较高,蓝色表示相对丰度较低),Group 代表不同的分组,左侧聚类树代表OTU(物种)的聚类。结果可以揭示不同的处理下丰度有着规律性变化的肠道微生物。
(三)
肠型分析Enterotype
2011年,EMBL(欧洲分子生物学实验室)的Peer Bork所领导的小组首次提出了肠型分析(Enterotype)概念。研究发现不同肠型(Enterotypes)并不受种族、性别、体重、健康程度或者年龄而影响,而主要在于细菌生态系统中酶的平衡状态,比如,“肠型1”中细菌大多为拟杆菌属(Bacteroides),有更多合成维生素B7(生物素)的酶,而“肠型2”大多为普氏菌属(Prevotella),更善于合成维生素B1(硫胺素)[6]。
随后关于肠型的研究层出不穷[7][8][9]。
肠型分析[6]利用JSD距离和Partition Around Medoids (PAM)聚类算法,基于物种相对丰度对样本进行聚类。使用Calinski-Harabasz指数对结果进行评估,以确定最佳聚类数。肠型可以通过聚类分析来可视化展示:

图片说明:上图中三种颜色代表三种不同的肠型,肠型是通过Jensen–Shannon距离对样本进行排序分类。图中每个点代表了一个样本,越相似的样本在图中距离越近,反之样本差异越大距离越远。
为了筛选聚类间具有显著差异的Biomarker,通过LEfSe分析的LDA(线性判别分析)实现降维并评估差异物种的影响大小,即得到LDA Score。 差异物种的LDA值分布图如下:

图片说明:LDA值分布柱状图中展示了LDA Score大于设定值(上图中默认设置为2),即组间具有统计学差异的 Biomarker,柱状图的长度代表差异贡献度大小(即LDA Score)。
每组中对差异贡献最大的物种即为该组所代表的肠型,并绘制boxplot图:
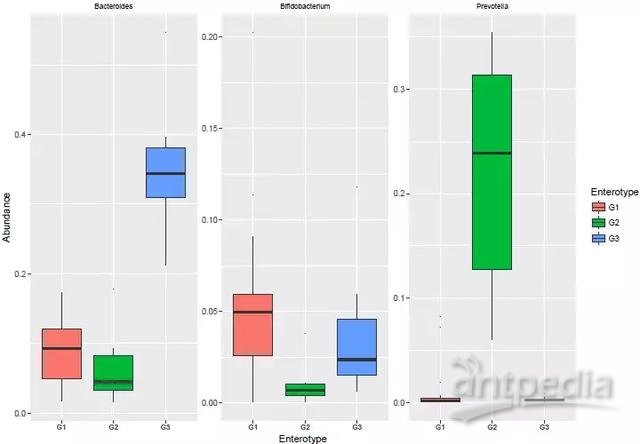
图片说明:箱线图说明了三种肠型上的三个标记属,不同颜色箱体代表不同肠型,四分位数的范围从第一个四分位数到第三个四分位数。中线表示中值。
参考文献
[1] Hehemann J H, Correc G, Barbeyron T, et al. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota.[J]. Nature, 2010, 464(7290):908-912.
[2] Olszak T, An D, Zeissig S, et al. Microbial Exposure During Early Life Has Persistent Effects on Natural Killer T Cell Function[J]. Inflammatory Bowel Disease Monitor, 2012, 336(6080):489-93.
[3] Peterson D A, Frank D N, Pace N R, et al. Metagenomic Approaches for Defining the Pathogenesis of Inflammatory Bowel Diseases[J]. Cell Host & Microbe, 2008, 3(6):417-427.
[4] Dicksved J, Halfvarson J, Rosenquist M, et al. Molecular analysis of the gut microbiota of identical twins with Crohn's disease.[J]. Isme Journal, 2008, 2(7):716-727.
[5] Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes.[J]. Nature, 2012, 490(7418):55-60.
[6] Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome.[J]. Nature, 2011, 473(7346):174.
[7] Wu G D, Chen J, Hoffmann C, et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes[J]. Science, 2011, 334(6052):105-8.
[8] Moeller A H, Degnan P H, Pusey A E, et al. Chimpanzees and Humans Harbor Compositionally Similar Gut Enterotypes[J]. Nature Communications, 2012, 3(6):1179.
[9] Wang J, Linnenbrink M, Künzel S, et al. Dietary history contributes to enterotype-like clustering and functional metagenomic content in the intestinal microbiome of wild mice.[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111(26):2703-10.
# END #
本文系欧易生物原创
转载请注明本文转自欧易生物
-
厂商文章
-
厂商文章

-
厂商文章

-
厂商文章
-
厂商文章

-
厂商文章

-
厂商文章

-
厂商文章

-
厂商文章
-
厂商文章

-
厂商文章