分享至
分享至



干货 | GSVA(Gene Set Variation Analysis)基因集变异分析


GSVA基因集变异分析,是在无参数且无监督的条件下,将Microarray和RNA-seq数据基因集进行富集的分析方法。GSVA能将一个基因-样本的数据矩阵(microarray data, FPKM, RPKM等)转换成基因集-样本矩阵。基于该矩阵可以进一步分析各个样本中基因集(如KEGG通路)的富集情况。由于GSVA的结果为一个基因集-样本的富集矩阵,相比其它基因集富集方法如GSEA (Gene Set Enrichment Analysis),下游分析会有更大的自由度。

在下游分析中,常见的分析是基于R包limma的差异分析。通过差异分析,找出实验组与对照组间存在富集值差异的通路。

准备:
R(推荐在RStudio上操作)、R包GSVA、GSEABase和limma
输入文件:
表达矩阵文件(FPKM、counts、RPKM等)和gmt矩阵文件(GSEA分析输入的通路矩阵文件)
表达矩阵文件示例:
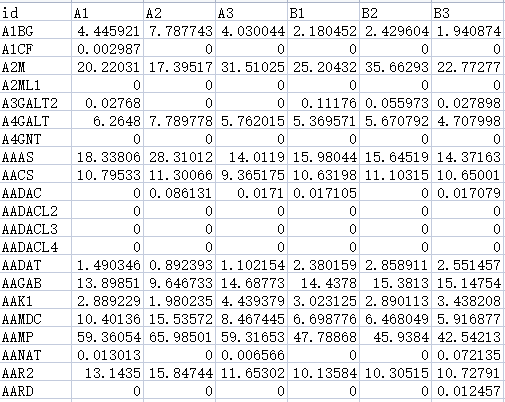
gmt矩阵文件示例:

代码示例
# 依赖包安装
install.packages("BiocManager")BiocManager::install("GSVA")library(''GSVA'')BiocManager::install("GSEABase")library(''GSEABase''
#输入fpkm.xls 表达矩阵文件
expset <- ''fpkm.xls''expset <- read.table(expset, sep = ''\\t'', header = TRUE, row.names = 1)
#Gaussian方法需要对表达矩阵log化
expset <- log2(expset + 1)matrix_expset <- data.matrix(expset)
# keg.backgroud.gmt gmt矩阵文件输入
gmt <- ''kegg.backgroud.gmt''geneSet <- getGmt(gmt)
# method可调整三种算法,针对不同需求,默认为gsva
kcdf参数包含不同统计方法,针对不同类型的输入矩阵应采用不同方法。与之同时纳入分析最小基因集,最大基因集也是可调的。输出结果将基因-样本的表达矩阵转换为通路-样本的富集分数矩阵。
gsva_scores <- gsva(matrix_expset, geneSet, method = ''gsva'', min.sz = 15 , max.sz = 5000, kcdf = ''Gaussian'', parallel.sz = 10)# 接着利用limma进行差异分析
BiocManager::install("limma")library(''limma'')groups <- factor(c(rep(''A'', 3,), rep(''B'',3)))design <- model.matrix(~ groups + 0)rownames(design) <- colnames(gsva_scores)compareE <- makeContrasts(groupsA-groupsB, levels = design)fit <- lmFit(gsva_scores, design)fit <- contrasts.fit(fit, compareE)fit <- eBayes(fit)
#得到未筛选的limma两组间比较结果
fit_all <- topTable(fit, coef = 1, adjust = ''BH'', n = Inf)fit_all <- cbind(rownames(fit_all),fit_all)colnames(fit_all) = c("geneset", "logFC", "avgExp", "t", "pval", "FDR", "B")
#于是便得到了一个gsva富集矩阵及组间的通路差异结果。
最后,可以对差异结果做一定的可视化,如热图或者柱状图。
#筛选部分结果
diff_result = fit_all[fit_all$pval < 0.05,]diff_result$up_down = ifelse(diff_result$t<0,''Down'',''Up'')
#做柱状图
library(ggplot2)pp = ggplot(diff_result, aes(reorder(geneset, t), t)) + geom_col(aes(fill=up_down)) + scale_fill_manual(values=c("#6CC570","#2A5078")) + coord_flip() +labs(x="Gene set", y="t value of GSVA Score" ) +theme_minimal() +geom_text(data = diff_result[diff_result$up_down == ''Down'',], aes(x= geneset, y=0, label = geneset), hjust = 0, size = 2.5)+geom_text(data = diff_result[diff_result$up_down == ''Up'',], aes(x= geneset, y=0, label = geneset), hjust = 1, size = 2.5)+ theme_bw() +theme(axis.line.x = element_line(colour = "black"))+theme(axis.line = element_blank()) +theme(axis.text.y=element_blank()) +theme(panel.grid =element_blank(),panel.border = element_blank(),axis.text.x=element_text(size=6),legend.text=element_text(size=8),axis.ticks = element_blank())+ labs( fill = "Up_Down")
#存图
ggsave("barplot.pdf",height = 10,width = 8,plot = pp, limitsize = FALSE)

- OE BIOTECH

OE BIOTECH -

END
排版人:小久
原创声明:本文由欧易生物(OEBIOTECH)学术团队报道,本文著作权归文章作者所有。欢迎个人转发及分享,未经作者的允许禁止转载。


点击“阅读全文” 收获更多精彩