分享至
分享至



您的质谱组学数据中存在批次效应吗?——氨探生物OBC工具助您全方位评估和校正

随着相关技术的成熟和普及,蛋白质组学和代谢组学开始在越来越多的临床队列研究中崭露头角。在蛋白质组和代谢组学的高通量实验中,实验人员需要在不同的时间处理大批量的样品,使用的试剂和前处理的条件可能会有差异;质谱仪在大批量样品分析的过程中,如遇色谱或者质谱的故障,则会中断分析,导致中断前后数据采集存在差异;此外,使用多台质谱仪同时分析样品,即使同一型号的质谱仪产出的数据也会有差异。这些可能引起样本间的系统性差异,称为批次效应(Batch effect)。批次效应对于数据分析和解释会带来极大挑战,它掩盖了真实的生物学差异,增加后续差异蛋白或者代谢物进一步验证和生物标志物开发的难度。而严格的质量控制和有效的数据处理方法是评估和校正批次效应的关键。
实验室质量控制能反映分析质量是否稳定,是质量控制的基础和核心。质控应尽量覆盖到每一位检测人员、每一台检测设备和每一类检测项目。举例而言,在样本前处理过程中,可以通过样品随机化、引入质控样本、进行标准的自动化前处理等措施来减少批次效应的引入(图1)。
· 样品随机化:通过随机分配样本到不同的实验批次中,减少批次效应。
· 质控样品:通过在每个批次中引入相同的质控样品(包括混合基质样本、标准品质控样本等),评估项目过程中的差异。

图1. 队列样品分析过程中的实验设计
然而,即便是经过严格的质量控制,批次效应仍然可能无法避免。因此,大队列组学数据都需要采用合适的方法预先评估数据的批次效应。常用的方法包括:强度分布(boxplot)、样品间的相关性(correlation)、主成分分析(PCA)、相对标准偏差(RSD)值、分层聚类(hierarchical clustering)、主成分方差分析(PVCA)等分析。
在确认数据中存在批次效应后,可以使用标准化或者归一化的方法使蛋白质或者代谢物的强度分布一致,如VSN(Variance Stabilizing Normalization)可以消除部分批次效应。在标准化或者归一化后,影响特定蛋白质或蛋白质组的批次效应可能仍然是方差的主要来源,需要更多算法和统计模型来校正批次效应,如HarmonizR, hRUV, ComBat等,这些方法可以更精确地调整批次间的差异,提高数据质量。
然而,批次效应评估和校正往往需要耗费大量时间和精力,需要从多个维度进行评判,而且需要在实验过程中实时评估数据质量,确定批次效应是否存在。为此,氨探生物设计了全新的web批次校正工具:Omics Batch Correct (图2),老师们可输入网址:https://omia.untangledbio.com/obc/或点击阅读原文直达。

图2. OBC校正工具首页
研究者只需要上传原始的蛋白定量或者代谢物定量结果,该工具可对组学数据进行归一化、自定义缺失比例的筛选、缺失值填充、多维度的批次评估与批次校正(图3)。为方便后续的生物信息学分析,所有的处理数据和图表均可下载。
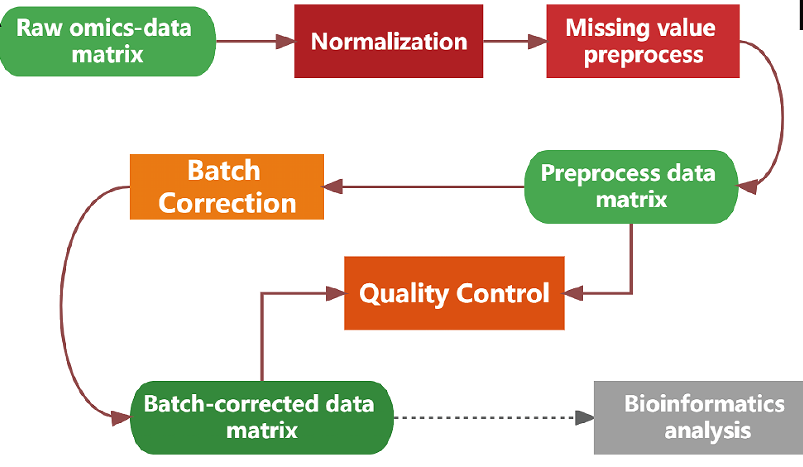
图3. OBC软件数据处理流程
OBC可以输出多种图表进行质控分析,供研究者评价数据质量,以下展示了5种典型的质控分析图:

图4. 典型的质控分析图
经过内部多个项目的项目数据集测试,我们发现无论是蛋白组学数据还是代谢组学数据,都能在OBC的帮助下进行高效、精确的处理。不再需要繁琐的手动操作和多种工具的组合,能够为您的组学研究提供更便捷、可靠的数据处理解决方案。
欢迎大家使用我们的工具,如果您在使用过程中遇到任何问题,可以随时和我们开发人员联系:helong.zheng@qlife-lab.com。
1.Poulos R C, Hains P G, Shah R, et al. Strategies to enable large-scale proteomics for reproducible research[J]. Nature communications, 2020, 11(1): 3793.
2.Voß H, Schlumbohm S, Barwikowski P, et al. HarmonizR enables data harmonization across independent proteomic datasets with appropriate handling of missing values[J]. Nature Communications, 2022, 13(1): 3523.
3.Välikangas T, Suomi T, Elo L L. A systematic evaluation of normalization methods in quantitative label-free proteomics[J]. Briefings in bioinformatics, 2018, 19(1): 1-11.
4.Johnson W E, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods[J]. Biostatistics, 2007, 8(1): 118-127.
5.Čuklina J, Lee CH, Williams EG, Sajic T, Collins BC, Rodríguez Martínez M, Sharma VS, Wendt F, Goetze S, Keele GR, Wollscheid B, Aebersold R, Pedrioli PGA. Diagnostics and correction of batch effects in large-scale proteomic studies: a tutorial. Mol Syst Biol. 2021 Aug;17(8):e10240.
解构健康奥秘、探寻生命答案,氨探生物以一流的分子表型组平台和成熟的临床转化应用体系,为优秀的研究团队进行技术和数据赋能,致力于实现分子表型水平的精准诊疗。