







当下最流行的表观遗传研究检测技术是什么?看看这里……
11月17日Cell杂志SnapShot专栏介绍了表观遗传研究的检测方法,这四种方法包括:亚硫酸氢钠测序法(bisulfite sequencing)、染色质免疫沉淀测序技术(chromatin immunoprecipiation sequencing)、开放染色质测定(determination of open chromatin),以及3D染色质捕获分析(3D chromatin capture)。
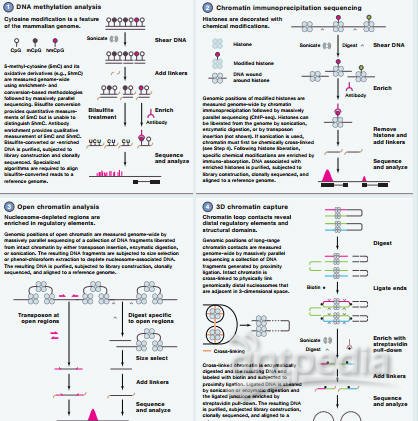
3D染色质捕获分析
虽然十年前我们就获得了人类基因组序列的线性图谱,但是对于其空间组织的破译,还才刚刚起步。最近的研究表明,远程基因组元件,如增强子和启动子,能通过染色质相互作用被带入其它区域,调控临近位置的基因转录。这打破了我们之前对于基因调控的理解,也指出了三维染色质结构的重要性。
近年来,染色质构象捕获(chromatin conformation capture)及更高通量的衍生技术的出现,有助于研究人员阐明染色质的复杂结构。这些技术提供了长距离的染色质相互作用,但不能扩展到整个染色质相互反应组。之后,马萨诸塞大学的Job Dekker开发出了HiC,将3C与新一代测序相结合,在全基因组范围内查看3D染色质相互作用。
尽管这些技术能提供染色质相互作用的全局视图,但近年来染色质捕获方法也许会有进一步的发展,分辨率更高,提供更为精细的染色质相互作用组图谱。如果这真的能实现,将有助于人们更深入地了解基因调控和人类疾病。
荷兰和以色列的研究组介绍了一种4C-seq技术,实现了前所未有的高分辨率,描绘染色质相互作用,为更准确,更细致的解析特殊功能元件的调控相互作用铺平了道路。
他们的方法首先与正常方法无异,也是联合消化和DNA连接两大步骤,这些连接后的DNA通常含有多个限制性片段,因此研究人员进行了又一轮的,不同限制性内切酶的消化,然后通过反向PCR扩增DNA目标区域特异性连接信号,最后利用新一代测序平台进行解析。
此外还有研究人员开发出了一个集成方案,可以在未知这些技术或生物来源的前提下,消除Hi-C数据中的许多误差。换句话说,通过一种无偏差方式消除了误差。其核心假设是,在一个无误差实验中,所有的基因组区域应该通过同一“可见性”进行分析,这样实验中观察到基因组区域的概率就相同了。而且作者假设每对相互作用的可见误差是会消失的,也就是说,这种偏差是随着相互作用的两个区域各自作用区域出现的误差而出现的。
在这些假设的基础上,Hi-C原始计数矩阵可以反复验证,在一个重复循环中,Hi-C图谱的每一组分都能通过两个互作区域的可见误差产物区分开来。研究人员也在人淋巴母细胞系实验中验证了这种方法,获得结果出现的误差与近期概率方法计算出来的限制性片段水平误差相关性很好,从而相互确认了这两种方法的作用。
利用开放染色质分析癌症
染色质是在细胞核中的材料,在细胞分裂过程中凝结形成染色体,是由DNA、被称为组蛋白的蛋白质和RNA组成的。每种类型的细胞都有一个特有的染色质结构,包括封闭的染色质——紧紧缠绕在核小体周围并且相对不活跃,和开放的染色质——宽松的材料片段,与编码在DNA中的调控元素相互作用。
美国Jackson实验室的研究人员发现了一种精确和可靠的方法——开放染色质的全基因组分析,来识别引起特定类型白血病的细胞,他们表示分析大量肿瘤细胞中的开放染色质,可以提供一种可能的改进方法,来识别癌细胞的起源,因为染色质结构有着细胞类型的特异性。
这项研究通过精通人类基因组学的计算机科学同事之间的密切合作,利用对我们来说崭新的先进基因组和基因组测序技术,迅速从这些技术中提取最大价值,将研究结果与人类基因组数据进行整合和比较,揭示最具有转化相关性的潜在生物学机制。
-
焦点事件

-
焦点事件

-
科技前沿

-
科技前沿

-
会议会展






